
Building things#
Quick links:
Simple bulk crystals:
bulk()Simple molecules:
molecule()Special surfaces:
fcc:
fcc100(),fcc110(),fcc111(),fcc211(),fcc111_root()bcc:
bcc100(),bcc110(),bcc111()* -bcc111_root()hcp:
hcp0001(),hcp10m10(),hcp0001_root()diamond:
diamond100(),diamond111()
\(MX_2\) (2H or 1T):
mx2()Other surface tools:
surface(),add_adsorbate(),add_vacuum(),root_surface()Other tools:
cut(),stack(),sort(),minimize_tilt(),niggli_reduce(),rotate(),minimize_rotation_and_translation(),get_deviation_from_optimal_cell_shape(),find_optimal_cell_shape(),make_supercell()Separation:
connected_indices(),connected_atoms(),separate(),split_bond()
See also
The
ase.latticemodule. The module contains functions for creating most common crystal structures with arbitrary orientation. The user can specify the desired Miller index along the three axes of the simulation, and the smallest periodic structure fulfilling this specification is created. Both bulk crystals and surfaces can be created.The
ase.clustermodule. Useful for creating nanoparticles and clusters.The
ase.spacegroupmoduleThe
ase.geometrymodule
Molecules#
The G2-database of common molecules is available:
- ase.build.molecule(name, vacuum=None, **kwargs)[source]#
Create an atomic structure from a database.
This is a helper function to easily create molecules from the g2 and extra databases.
- Parameters:
- Returns:
An ASE Atoms object corresponding to the specified molecule.
- Return type:
Notes
To see a list of allowed names, try:
>>> from ase.collections import g2 >>> print(g2.names) ['PH3', 'P2', 'CH3CHO', 'H2COH', 'CS', 'OCHCHO', 'C3H9C', 'CH3COF', 'CH3CH2OCH3', 'HCOOH', 'HCCl3', 'HOCl', 'H2', 'SH2', 'C2H2', 'C4H4NH', 'CH3SCH3', 'SiH2_s3B1d', 'CH3SH', 'CH3CO', 'CO', 'ClF3', 'SiH4', 'C2H6CHOH', 'CH2NHCH2', 'isobutene', 'HCO', 'bicyclobutane', 'LiF', 'Si', 'C2H6', 'CN', 'ClNO', 'S', 'SiF4', 'H3CNH2', 'methylenecyclopropane', 'CH3CH2OH', 'F', 'NaCl', 'CH3Cl', 'CH3SiH3', 'AlF3', 'C2H3', 'ClF', 'PF3', 'PH2', 'CH3CN', 'cyclobutene', 'CH3ONO', 'SiH3', 'C3H6_D3h', 'CO2', 'NO', 'trans-butane', 'H2CCHCl', 'LiH', 'NH2', 'CH', 'CH2OCH2', 'C6H6', 'CH3CONH2', 'cyclobutane', 'H2CCHCN', 'butadiene', 'C', 'H2CO', 'CH3COOH', 'HCF3', 'CH3S', 'CS2', 'SiH2_s1A1d', 'C4H4S', 'N2H4', 'OH', 'CH3OCH3', 'C5H5N', 'H2O', 'HCl', 'CH2_s1A1d', 'CH3CH2SH', 'CH3NO2', 'Cl', 'Be', 'BCl3', 'C4H4O', 'Al', 'CH3O', 'CH3OH', 'C3H7Cl', 'isobutane', 'Na', 'CCl4', 'CH3CH2O', 'H2CCHF', 'C3H7', 'CH3', 'O3', 'P', 'C2H4', 'NCCN', 'S2', 'AlCl3', 'SiCl4', 'SiO', 'C3H4_D2d', 'H', 'COF2', '2-butyne', 'C2H5', 'BF3', 'N2O', 'F2O', 'SO2', 'H2CCl2', 'CF3CN', 'HCN', 'C2H6NH', 'OCS', 'B', 'ClO', 'C3H8', 'HF', 'O2', 'SO', 'NH', 'C2F4', 'NF3', 'CH2_s3B1d', 'CH3CH2Cl', 'CH3COCl', 'NH3', 'C3H9N', 'CF4', 'C3H6_Cs', 'Si2H6', 'HCOOCH3', 'O', 'CCH', 'N', 'Si2', 'C2H6SO', 'C5H8', 'H2CF2', 'Li2', 'CH2SCH2', 'C2Cl4', 'C3H4_C3v', 'CH3COCH3', 'F2', 'CH4', 'SH', 'H2CCO', 'CH3CH2NH2', 'Li', 'N2', 'Cl2', 'H2O2', 'Na2', 'BeH', 'C3H4_C2v', 'NO2'] >>> from ase.build.molecule import extra >>> print(extra.keys()) dict_keys(['Be2', 'C7NH5', 'BDA', 'biphenyl', 'C60'])
Examples
>>> from ase.build import molecule >>> atoms = molecule('H2O')
Example:
>>> from ase.build import molecule
>>> atoms = molecule('H2O')
The list of available molecules is those from the ase.collections.g2
database:
>>> from ase.collections import g2
>>> g2.names
['PH3', 'P2', 'CH3CHO', 'H2COH', 'CS', 'OCHCHO', 'C3H9C', 'CH3COF',
'CH3CH2OCH3', 'HCOOH', 'HCCl3', 'HOCl', 'H2', 'SH2', 'C2H2',
'C4H4NH', 'CH3SCH3', 'SiH2_s3B1d', 'CH3SH', 'CH3CO', 'CO', 'ClF3',
'SiH4', 'C2H6CHOH', 'CH2NHCH2', 'isobutene', 'HCO', 'bicyclobutane',
'LiF', 'Si', 'C2H6', 'CN', 'ClNO', 'S', 'SiF4', 'H3CNH2',
'methylenecyclopropane', 'CH3CH2OH', 'F', 'NaCl', 'CH3Cl',
'CH3SiH3', 'AlF3', 'C2H3', 'ClF', 'PF3', 'PH2', 'CH3CN',
'cyclobutene', 'CH3ONO', 'SiH3', 'C3H6_D3h', 'CO2', 'NO',
'trans-butane', 'H2CCHCl', 'LiH', 'NH2', 'CH', 'CH2OCH2',
'C6H6', 'CH3CONH2', 'cyclobutane', 'H2CCHCN', 'butadiene', 'C',
'H2CO', 'CH3COOH', 'HCF3', 'CH3S', 'CS2', 'SiH2_s1A1d', 'C4H4S',
'N2H4', 'OH', 'CH3OCH3', 'C5H5N', 'H2O', 'HCl', 'CH2_s1A1d',
'CH3CH2SH', 'CH3NO2', 'Cl', 'Be', 'BCl3', 'C4H4O', 'Al', 'CH3O',
'CH3OH', 'C3H7Cl', 'isobutane', 'Na', 'CCl4', 'CH3CH2O', 'H2CCHF',
'C3H7', 'CH3', 'O3', 'P', 'C2H4', 'NCCN', 'S2', 'AlCl3', 'SiCl4',
'SiO', 'C3H4_D2d', 'H', 'COF2', '2-butyne', 'C2H5', 'BF3', 'N2O',
'F2O', 'SO2', 'H2CCl2', 'CF3CN', 'HCN', 'C2H6NH', 'OCS', 'B', 'ClO',
'C3H8', 'HF', 'O2', 'SO', 'NH', 'C2F4', 'NF3', 'CH2_s3B1d', 'CH3CH2Cl',
'CH3COCl', 'NH3', 'C3H9N', 'CF4', 'C3H6_Cs', 'Si2H6', 'HCOOCH3', 'O',
'CCH', 'N', 'Si2', 'C2H6SO', 'C5H8', 'H2CF2', 'Li2', 'CH2SCH2', 'C2Cl4',
'C3H4_C3v', 'CH3COCH3', 'F2', 'CH4', 'SH', 'H2CCO', 'CH3CH2NH2', 'Li',
'N2', 'Cl2', 'H2O2', 'Na2', 'BeH', 'C3H4_C2v', 'NO2']
plus Be2, C7NH5, BDA, biphenyl and C60 (for historical
reasons).
More complicated molecules may be obtained using the PubChem API integration in
the pubchem_atoms_search() and pubchem_atoms_conformer_search()
functions. You may search based on common name, chemical identification number
(cid), smiles string, or conformer identification number.
Common bulk crystals#
- ase.build.bulk(name: str, crystalstructure: str = None, a: float = None, b: float = None, c: float = None, *, alpha: float = None, covera: float = None, u: float = None, orthorhombic: bool = False, cubic: bool = False, basis=None) Atoms[source]#
Creating bulk systems.
Crystal structure and lattice constant(s) will be guessed if not provided.
- name: str
Chemical symbol or symbols as in ‘MgO’ or ‘NaCl’.
- crystalstructure: str
Must be one of sc, fcc, bcc, tetragonal, bct, hcp, rhombohedral, orthorhombic, mcl, diamond, zincblende, rocksalt, cesiumchloride, fluorite or wurtzite.
- a: float
Lattice constant.
- b: float
Lattice constant. If only a and b is given, b will be interpreted as c instead.
- c: float
Lattice constant.
- alpha: float
Angle in degrees for rhombohedral lattice.
- covera: float
c/a ratio used for hcp. Default is ideal ratio: sqrt(8/3).
- u: float
Internal coordinate for Wurtzite structure.
- orthorhombic: bool
Construct orthorhombic unit cell instead of primitive cell which is the default.
- cubic: bool
Construct cubic unit cell if possible.
examples:
>>> from ase.build import bulk
>>> a1 = bulk('Cu', 'fcc', a=3.6)
>>> a2 = bulk('Cu', 'fcc', a=3.6, orthorhombic=True)
>>> a3 = bulk('Cu', 'fcc', a=3.6, cubic=True)
>>> a1.cell
array([[ 0. , 1.8, 1.8],
[ 1.8, 0. , 1.8],
[ 1.8, 1.8, 0. ]])
>>> a2.cell
array([[ 2.546, 0. , 0. ],
[ 0. , 2.546, 0. ],
[ 0. , 0. , 3.6 ]])
>>> a3.cell
array([[ 3.6, 0. , 0. ],
[ 0. , 3.6, 0. ],
[ 0. , 0. , 3.6]])



Nanotubes#
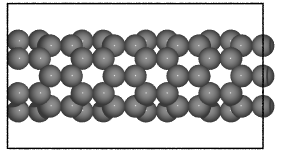
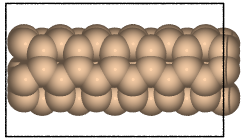
- ase.build.nanotube(n, m, length=1, bond=1.42, symbol='C', verbose=False, vacuum=None)[source]#
Create an atomic structure.
Creates a single-walled nanotube whose structure is specified using the standardized (n, m) notation.
- Parameters:
n (int) – n in the (n, m) notation.
m (int) – m in the (n, m) notation.
length (int, optional) – Length (axial repetitions) of the nanotube.
bond (float, optional) – Bond length between neighboring atoms.
symbol (str, optional) – Chemical element to construct the nanotube from.
verbose (bool, optional) – If True, will display key geometric parameters.
- Returns:
An ASE Atoms object corresponding to the specified molecule.
- Return type:
Examples
>>> from ase.build import nanotube >>> atoms1 = nanotube(6, 0, length=4) >>> atoms2 = nanotube(3, 3, length=6, bond=1.4, symbol='Si')
examples:
>>> from ase.build import nanotube
>>> cnt1 = nanotube(6, 0, length=4)
>>> cnt2 = nanotube(3, 3, length=6, bond=1.4, symbol='Si')
Graphene nanoribbons#
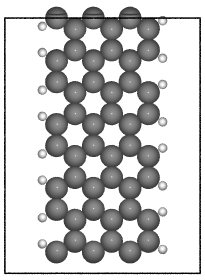
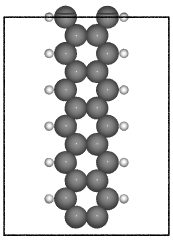
- ase.build.graphene_nanoribbon(n, m, type='zigzag', saturated=False, C_H=1.09, C_C=1.42, vacuum=None, magnetic=False, initial_mag=1.12, sheet=False, main_element='C', saturate_element='H')[source]#
Create a graphene nanoribbon.
Creates a graphene nanoribbon in the x-z plane, with the nanoribbon running along the z axis.
Parameters:
- n: int
The width of the nanoribbon. For armchair nanoribbons, this n may be half-integer to repeat by half a cell.
- m: int
The length of the nanoribbon.
- type: str
The orientation of the ribbon. Must be either ‘zigzag’ or ‘armchair’.
- saturated: bool
If true, hydrogen atoms are placed along the edge.
- C_H: float
Carbon-hydrogen bond length. Default: 1.09 Angstrom.
- C_C: float
Carbon-carbon bond length. Default: 1.42 Angstrom.
- vacuum: None (default) or float
Amount of vacuum added to non-periodic directions, if present.
- magnetic: bool
Make the edges magnetic.
- initial_mag: float
Magnitude of magnetic moment if magnetic.
- sheet: bool
If true, make an infinite sheet instead of a ribbon (default: False)
examples:
>>> from ase.build import graphene_nanoribbon
>>> gnr1 = graphene_nanoribbon(3, 4, type='armchair', saturated=True,
vacuum=3.5)
>>> gnr2 = graphene_nanoribbon(2, 6, type='zigzag', saturated=True,
... C_H=1.1, C_C=1.4, vacuum=3.0,
... magnetic=True, initial_mag=1.12)
ASE contains a number of modules for setting up atomic structures, mainly molecules, bulk crystals and surfaces. Some of these modules have overlapping functionality, but strike a different balance between flexibility and ease-of-use.
Attaching structures#
- ase.build.attach.attach(atoms1, atoms2, distance, direction=(1, 0, 0), maxiter=50, accuracy=1e-05)[source]#
Attach two structures
- Parameters:
atoms1 (Atoms) – cell and pbc of this object are used
atoms2 (Atoms)
distance (float) – minimal distance (Angstrom)
direction (unit vector (3 floats)) – relative direction between center of masses
maxiter (int) – maximal number of iterations to get required distance, default 100
accuracy (float) – required accuracy for minimal distance (Angstrom), default 1e-5
- Return type:
Joined structure as an atoms object.
- ase.build.attach.attach_randomly(atoms1, atoms2, distance, rng=<module 'numpy.random' from '/scratch/jensj/ase-docs/venv/lib/python3.11/site-packages/numpy/random/__init__.py'>)[source]#
Randomly attach two structures with a given minimal distance
- Parameters:
atoms1 (Atoms object)
atoms2 (Atoms object)
distance (float) – Required distance
rng (random number generator object) – defaults to np.random.RandomState()
- Return type:
Joined structure as an atoms object.
- ase.build.attach.attach_randomly_and_broadcast(atoms1, atoms2, distance, rng=<module 'numpy.random' from '/scratch/jensj/ase-docs/venv/lib/python3.11/site-packages/numpy/random/__init__.py'>, comm=<ase.parallel.MPI object>)[source]#
- Randomly attach two structures with a given minimal distance
and ensure that these are distributed.
- Parameters:
atoms1 (Atoms object)
atoms2 (Atoms object)
distance (float) – Required distance
rng (random number generator object) – defaults to np.random.RandomState()
comm (communicator to distribute) – Communicator to distribute the structure, default: world
- Return type:
Joined structure as an atoms object.