
Note#
A newer implementation of the Debye Scattering Equation can be found at FrederikLizakJohansen/DebyeCalculator which supports more atomic elements, neutron scattering, calculation of pair distribution function data and it is significantly faster on CPU while supporting GPU acceleration. The new implementation can take ase objects as input.
X-ray scattering simulation#
The module for simulation of X-ray scattering properties from the atomic level. The approach works only for finite systems, so that periodic boundary conditions and cell shape are ignored.
Theory#
The scattering can be calculated using Debye formula [Debye1915] :
where:
\(a\) and \(b\) – atom indexes;
\(f_a(q)\) – \(a\)-th atomic scattering factor;
\(r_{ab}\) – distance between atoms \(a\) and \(b\);
\(q\) is a scattering vector length defined using scattering angle (\(\theta\)) and wavelength (\(\lambda\)) as \(q = 4\pi \cdot \sin(\theta)/\lambda\).
The thermal vibration of atoms can be accounted by introduction of damping exponent factor (Debye-Waller factor) written as \(\exp(-B \cdot q^2 / 2)\). The angular dependency of geometrical and polarization factors are expressed as [Iwasa2007] \(\cos(\theta)/(1 + \alpha \cos^2(2\theta))\), where \(\alpha \approx 1\) if incident beam is not polarized.
Units#
The following measurement units are used:
scattering vector \(q\) – inverse Angstrom (1/Å),
thermal damping parameter \(B\) – squared Angstrom (Å2).
Example#
The considered system is a nanoparticle of silver which is built using
FaceCenteredCubic function (see ase.cluster) with parameters
selected to produce approximately 2 nm sized particle:
from ase.cluster.cubic import FaceCenteredCubic
from ase.utils.xrdebye import XrDebye
import numpy as np
surfaces = [(1, 0, 0), (1, 1, 0), (1, 1, 1)]
atoms = FaceCenteredCubic('Ag', [(1, 0, 0), (1, 1, 0), (1, 1, 1)],
[6, 8, 8], 4.09)
Next, we need to specify the wavelength of the X-ray source:
xrd = XrDebye(atoms=atoms, wavelength=0.50523)
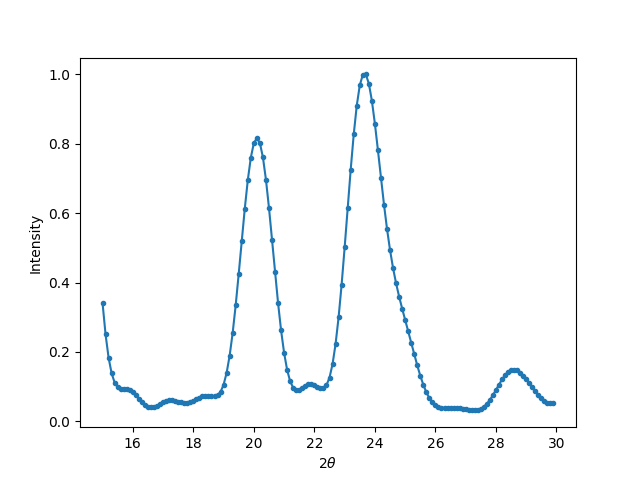
The X-ray diffraction pattern on the \(2\theta\) angles ranged from 15 to 30 degrees can be simulated as follows:
xrd.calc_pattern(x=np.arange(15, 30, 0.1), mode='XRD')
xrd.plot_pattern('xrd.png')
The resulted X-ray diffraction pattern shows (220) and (311) peaks at 20 and ~24 degrees respectively.
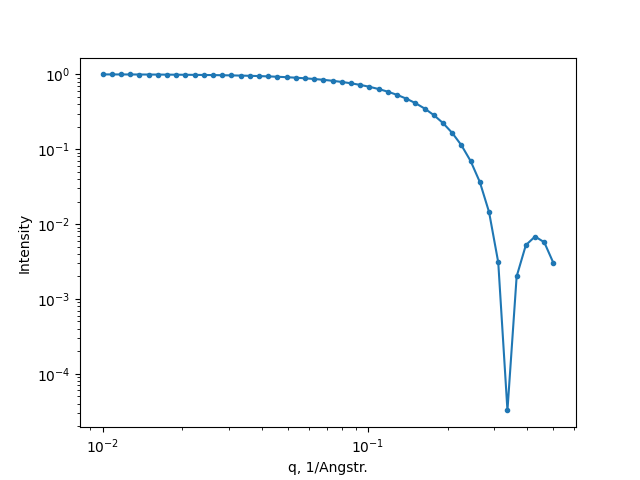
The small-angle scattering curve can be simulated too. Assuming that scattering vector is ranged from \(10^{-2}=0.01\) to \(10^{-0.3}\approx 0.5\) 1/Å the following code should be run:
xrd.calc_pattern(x=np.logspace(-2, -0.3, 50), mode='SAXS')
xrd.plot_pattern('saxs.png')
The resulted SAXS pattern:
Further details#
The module contains wavelengths dictionary with X-ray wavelengths for copper and wolfram anodes:
from ase.utils.xrdebye import wavelengths
print('Cu Kalpha1 wavelength: %f Angstr.' % wavelengths['CuKa1'])
The dependence of atomic form-factors from scattering vector is calculated
based on coefficients given in waasmaier dictionary according
[Waasmaier1995] if method of calculations is set to ‘Iwasa’. In other case,
the atomic factor is equal to atomic number and angular damping factor is
omitted.
XrDebye class members#
- class ase.utils.xrdebye.XrDebye(atoms, wavelength, damping=0.04, method='Iwasa', alpha=1.01, warn=True)[source]#
Class for calculation of XRD or SAXS patterns.
Initilize the calculation of X-ray diffraction patterns
Parameters:
- atoms: ase.Atoms
atoms object for which calculation will be performed.
- wavelength: float, Angstrom
X-ray wavelength in Angstrom. Used for XRD and to setup dumpings.
- dampingfloat, Angstrom**2
thermal damping factor parameter (B-factor).
- method: {‘Iwasa’}
method of calculation (damping and atomic factors affected).
If set to ‘Iwasa’ than angular damping and q-dependence of atomic factors are used.
For any other string there will be only thermal damping and constant atomic factors (\(f_a(q) = Z_a\)).
- alpha: float
parameter for angular damping of scattering intensity. Close to 1.0 for unplorized beam.
- warn: boolean
flag to show warning if atomic factor can’t be calculated
- calc_pattern(x=None, mode='XRD', verbose=False)[source]#
Calculate X-ray diffraction pattern or small angle X-ray scattering pattern.
Parameters:
- x: float array
points where intensity will be calculated. XRD - 2theta values, in degrees; SAXS - q values in 1/A (\(q = 2 \pi \cdot s = 4 \pi \sin( \theta) / \lambda\)). If
xisNonethen default values will be used.- mode: {‘XRD’, ‘SAXS’}
the mode of calculation: X-ray diffraction (XRD) or small-angle scattering (SAXS).
- Returns:
list of intensities calculated for values given in
x.
- get(s)[source]#
Get the powder x-ray (XRD) scattering intensity using the Debye-Formula at single point.
Parameters:
- s: float, in inverse Angstrom
scattering vector value (\(s = q / 2\pi\)).
- Returns:
Intensity at given scattering vector \(s\).
- get_waasmaier(symbol, s)[source]#
Scattering factor for free atoms.
Parameters:
- symbol: string
atom element symbol.
- s: float, in inverse Angstrom
scattering vector value (\(s = q / 2\pi\)).
- Returns:
Intensity at given scattering vector \(s\).
Note
for hydrogen will be returned zero value.
References#
Debye Ann. Phys. 351, 809–823 (1915)
Iwasa, K. Nobusada J. Phys. Chem. C, 111, 45-49 (2007) doi: 10.1021/jp063532w
Waasmaier, A. Kirfel Acta Cryst. A51, 416-431 (1995)