
The data module#
Atomic data#
This module defines the following variables:
- ase.data.atomic_masses#
- ase.data.atomic_names#
- ase.data.chemical_symbols#
- ase.data.covalent_radii#
- ase.data.cpk_colors#
- ase.data.reference_states#
- ase.data.vdw_radii#
- ase.data.atomic_masses_iupac2016#
- ase.data.atomic_masses_legacy#
- ase.data.cohesive_energies#
All of these are lists that should be indexed with an atomic number:
>>> from ase.data import atomic_numbers, atomic_names, atomic_masses, covalent_radii
>>> atomic_names[92]
'Uranium'
>>> atomic_masses[2]
4.0026000000000002
- ase.data.atomic_numbers#
If you don’t know the atomic number of some element, then you can look
it up in the atomic_numbers dictionary:
>>> atomic_numbers['Cu']
29
>>> covalent_radii[29]
1.3200000000000001
Atomic masses are based on [Meija2016] (same array as
atomic_masses_iupac2016).
Standard atomic weights are taken from Table 1: “Standard atomic weights 2013”, with the uncertainties ignored.
For hydrogen, helium, boron, carbon, nitrogen, oxygen, magnesium, silicon, sulfur, chlorine, bromine and thallium, where the weights are given as a range the “conventional” weights are taken from Table 3, and the ranges are given in the source code comments.
The mass of the most stable isotope (in Table 4) is used for elements where there the element has no stable isotopes (to avoid NaNs): Tc, Pm, Po, At, Rn, Fr, Ra, Ac, everything after Np
Atomic masses provided by ASE before 2017 can be accessed in the
atomic_masses_legacy member. To recover legacy behaviour an
Atoms object can be modified as:
>>> from ase.data import atomic_masses_legacy
>>> atoms.set_masses(atomic_masses_legacy[atoms.numbers])
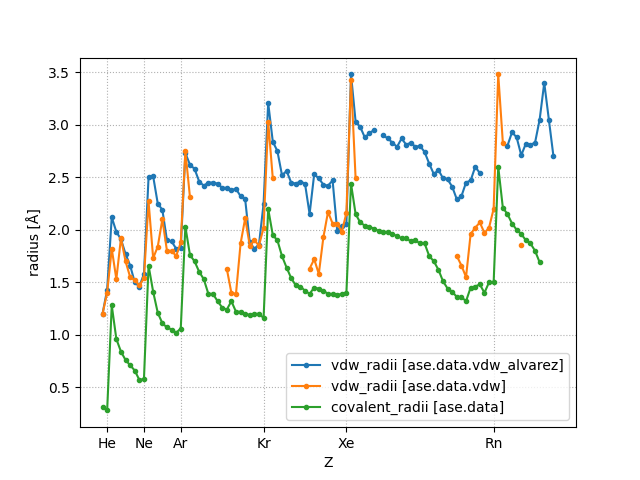
The covalent radii are taken from [Cordeo08].
The source of the van der Waals radii is given in vdw.py.
A newer source of van der Waals radii is given in vdw_alvarez.py. These radii are taken from [Alvarez13].
Atomic weights of the elements 2013 (IUPAC Technical Report). Meija, J., Coplen, T., Berglund, M., et al. (2016). Pure and Applied Chemistry, 88(3), pp. 265-291. Retrieved 30 Nov. 2016, from doi: 10.1515/pac-2015-0305
Covalent radii revisited, Beatriz Cordero, Verónica Gómez, Ana E. Platero-Prats, Marc Revés, Jorge Echeverría, Eduard Cremades, Flavia Barragán and Santiago Alvarez, Dalton Trans., 2008, 2832-2838 doi: 10.1039/B801115J
A cartography of the van der Waals territories, Alvarez, S., Dalton Trans., 2013, 42, 8617-8636, doi: 10.1039/C3DT50599E
How to extract isotope data from NIST#
- ase.data.isotopes.download_isotope_data()[source]#
Download isotope data from NIST public website.
Relative atomic masses of individual isotopes their abundance (mole fraction) are compiled into a dictionary. Individual items can be indexed by the atomic number and mass number, e.g. titanium-48:
>>> from ase.data.isotopes import download_isotope_data >>> isotopes = download_isotope_data() >>> isotopes[22][48]['mass'] 47.94794198 >>> isotopes[22][48]['composition'] 0.7372
Molecular data#
The G1, G2, and G3-databases are available. Example:
>>> from ase.build import molecule
>>> atoms = molecule('H2O')
All molecular members of each database is conveniently contained in a list of strings (g1, g2, g3), ??? and one can look up the experimental atomization energy for each molecule. This is extrapolated from experimental heats of formation at room temperature, using calculated zero-point energies and thermal corrections.
Example:
>>> from ase.data.g2 import get_atomization_energy
>>> get_atomization_energy('H2O')
232.5799
>>> from ase.units import kcal,mol
>>> get_atomization_energy('H2O')*kcal/mol
10.08561894878958
where the last line converts the experimental atomization energy of H2O from units of kcal/mol to eV.
Structures for compounds not found in the G1, G2, and G3-databases can
be obtained using the PubChem API integration in the
pubchem.pubchem_atoms_search() and
pubchem.pubchem_atoms_conformer_search() functions. You may
search based on common name, chemical identification number(cid),
smiles string, or conformer identification number.
- ase.data.pubchem.pubchem_atoms_search(*args, **kwargs)[source]#
Search PubChem for the field and search input on the argument passed in returning an atoms object.Note that only one argument may be passed in at a time.
- Parameters:
ase.data.pubchem.pubchem_search (see)
- Returns:
an ASE Atoms object containing the information on the requested entry
- Return type:
atoms (ASE Atoms Object)
- ase.data.pubchem.pubchem_atoms_conformer_search(*args, **kwargs)[source]#
Search PubChem for all the conformers of a given compound. Note that only one argument may be passed in at a time.
- Parameters:
ase.data.pubchem.pubchem_search (see)
- Returns:
a list containing the atoms objects of all the conformers for your search
- Return type:
conformers (list)
examples:
>>> from ase.data.pubchem import pubchem_atoms_search, pubchem_atoms_conformer_search
>>> cumene = pubchem_atoms_search(name='cumene')
>>> benzene = pubchem_atoms_search(cid=241)
>>> ethanol = pubchem_atoms_search(smiles='CCOH')
>>> octane_conformers = pubchem_atoms_conformer_search(name='octane')
To get all the data available on Pubchem use pubchem.pubchem_search() and
pubchem.pubchem_conformer_search().
- ase.data.pubchem.pubchem_search(*args, **kwargs) PubchemData[source]#
Search PubChem for the field and search input on the argument passed in returning a PubchemData object. Note that only one argument may be passed in at a time.
- Parameters:
- Returns:
a pubchem data object containing the information on the requested entry
- Return type:
result (PubchemData)
- ase.data.pubchem.pubchem_conformer_search(*args, **kwargs) list[source]#
Search PubChem for all the conformers of a given compound. Note that only one argument may be passed in at a time.
- Parameters:
ase.data.pubchem.pubchem_search (see)
- Returns:
a list containing the PubchemData objects of all the conformers for your search
- Return type:
conformers (list)
S22, s26, and s22x5 data#
The s22, s26, and s22x5 databases are available in the s22 module.
Each weakly bonded complex is identified as an entry in a list of strings (s22, s26, s22x5), and is fully created by a ‘create’-function:
>>> from ase.data.s22 import s22, create_s22_system
>>> sys = s22[0]
>>> sys
'Ammonia_dimer'
>>> atoms = create_s22_system(sys)
>>> atoms.get_chemical_symbols()
['N', 'H', 'H', 'H', 'N', 'H', 'H', 'H']
The coupled-cluster interaction energies for the s22 and s26 systems are retrieved like this:
>>> from ase.data.s22 import s22, get_interaction_energy_s22
>>> get_interaction_energy_s22(s22[0])
-0.1375
in units of eV. For s22 these are not the original energies, but from more recent work where the same (large) basis set was used for all complexes, yielding more accurate coupled-cluster interaction energies.
The s22x5 database expands on the original s22 data by introducing non-equilibrium geometries for each complex (0.9, 1.0, 1.2, 1.5, and 2.0 times original intermolecular distance). However, these calculations were done in accordance with the methods used in the original s22 work, and so is expected to inherit the same problems with mixed basis set sizes. Assuming the interaction energy error due to this is the same in all 5 geometries for each complex, the default s22x5 interaction energies are therefore corrected with the energy difference between original and newer energies at the original separation.
Example:
>>> from ase.data.s22 import *
>>> sys1 = s22[0]
>>> sys1
'Ammonia_dimer'
>>> atoms1 = create_s22_system(sys1)
>>> sys2 = s22x5[0]
>>> sys2
'Ammonia_dimer_0.9'
>>> atoms2 = create_s22_system(sys2)
>>> sys3 = s22x5[1]
>>> sys3
'Ammonia_dimer_1.0'
>>> atoms3 = create_s22_system(sys3)
>>> get_interaction_energy_s22(sys1)
-0.1375
>>> get_interaction_energy_s22(sys2)
-0.1375
>>> get_interaction_energy_s22(sys3)
-0.1375
>>> get_interaction_energy_s22x5(sys2)
-0.10549743024963291
>>> get_interaction_energy_s22x5(sys3)
-0.1375
>>> get_interaction_energy_s22x5(sys3,correct_offset=False)
-0.1362
>>> get_interaction_energy_s22x5(sys1,dist=1.0)
-0.1375
>>> get_interaction_energy_s22x5(sys1,dist=0.9)
-0.10549743024963291
>>> get_interaction_energy_s22x5(sys1,dist=0.9,correct_offset=False)
-0.1045
>>> get_number_of_dimer_atoms(sys1)
[4, 4]
>>> get_s22x5_distance(sys2)
-0.25040236345454536
>>> get_s22x5_distance(sys3)
0.0
where sys1 is an s22 complex in the original geometry, while sys2 and sys3 are two different s22x5 geometries of the same complex. It is seen that the interaction energies for an s22 system and its s22x5 equivalent (indexed ‘_1.0’) does not necessarily match when the energy offset-correction is turned off. The last two functions are convenience functions, giving the number of atoms in the two molecules constituting a dimer and the relative intermolecular distance in a dimer (relative to the ‘1.0’ separation, and in Angstrom), respectively.