
Using the spacegroup subpackage#
The most evident usage of the spacegroup subpackage is to set up an initial unit of a bulk structure. For this you only need to supply the unique atoms and their scaled positions, space group and lattice parameters.
Examples of setting up bulk structures#
We start by showing some examples of how to set up some common or
interesting bulk structures using
ase.spacegroup.crystal(). This function takes a lot of
arguments:
- ase.spacegroup.crystal(symbols=None, basis=None, occupancies=None, spacegroup=1, setting=1, cell=None, cellpar=None, ab_normal=(0, 0, 1), a_direction=None, size=(1, 1, 1), onduplicates='warn', symprec=0.001, pbc=True, primitive_cell=False, **kwargs) Atoms[source]#
Create an Atoms instance for a conventional unit cell of a space group.
Parameters:
- symbolsstr | sequence of str | sequence of Atom | Atoms
Element symbols of the unique sites. Can either be a string formula or a sequence of element symbols. E.g. (‘Na’, ‘Cl’) and ‘NaCl’ are equivalent. Can also be given as a sequence of Atom objects or an Atoms object.
- basislist of scaled coordinates
Positions of the unique sites corresponding to symbols given either as scaled positions or through an atoms instance. Not needed if symbols is a sequence of Atom objects or an Atoms object.
- occupancieslist of site occupancies
Occupancies of the unique sites. Defaults to 1.0 and thus no mixed occupancies are considered if not explicitly asked for. If occupancies are given, the most dominant species will yield the atomic number. The occupancies in the atoms.info[‘occupancy’] dictionary will have integers keys converted to strings. The conversion is done in order to avoid unexpected conversions when using the JSON serializer.
- spacegroupint | string | Spacegroup instance
Space group given either as its number in International Tables or as its Hermann-Mauguin symbol.
- setting1 | 2
Space group setting.
- cell3x3 matrix
Unit cell vectors.
- cellpar[a, b, c, alpha, beta, gamma]
Cell parameters with angles in degree. Is not used when \(cell\) is given.
- ab_normalvector
Is used to define the orientation of the unit cell relative to the Cartesian system when \(cell\) is not given. It is the normal vector of the plane spanned by a and b.
- a_directionvector
Defines the orientation of the unit cell a vector. a will be parallel to the projection of \(a_direction\) onto the a-b plane.
- size3 positive integers
How many times the conventional unit cell should be repeated in each direction.
- onduplicates‘keep’ | ‘replace’ | ‘warn’ | ‘error’
- Action if \(basis\) contain symmetry-equivalent positions:
‘keep’ - ignore additional symmetry-equivalent positions ‘replace’ - replace ‘warn’ - like ‘keep’, but issue an UserWarning ‘error’ - raises a SpacegroupValueError
- symprecfloat
Minimum “distance” betweed two sites in scaled coordinates before they are counted as the same site.
- pbcone or three bools
Periodic boundary conditions flags. Examples: True, False, 0, 1, (1, 1, 0), (True, False, False). Default is True.
- primitive_cellbool
Whether to return the primitive instead of the conventional unit cell.
Keyword arguments:
All additional keyword arguments are passed on to the Atoms constructor. Currently, probably the most useful additional keyword arguments are \(info\), \(constraint\) and \(calculator\).
Examples:
Two diamond unit cells (space group number 227)
>>> diamond = crystal('C', [(0,0,0)], spacegroup=227, ... cellpar=[3.57, 3.57, 3.57, 90, 90, 90], size=(2,1,1)) >>> ase.view(diamond)
A CoSb3 skutterudite unit cell containing 32 atoms
>>> skutterudite = crystal(('Co', 'Sb'), ... basis=[(0.25,0.25,0.25), (0.0, 0.335, 0.158)], ... spacegroup=204, cellpar=[9.04, 9.04, 9.04, 90, 90, 90]) >>> len(skutterudite) 32
There is also a get_spacegroup() function that will return a spacegroup object from an
Atoms object.
Aluminium (fcc)#
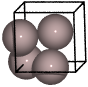
from ase.spacegroup import crystal
a = 4.05
al = crystal('Al', [(0, 0, 0)], spacegroup=225, cellpar=[a, a, a, 90, 90, 90])
The spacegroup argument can also be entered with its Hermann-Mauguin symbol, e.g. spacegroup=225 is equivalent to spacegroup=’F m -3 m’.
Iron (bcc)#
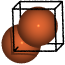
from ase.spacegroup import crystal
a = 2.87
fe = crystal('Fe', [(0, 0, 0)], spacegroup=229, cellpar=[a, a, a, 90, 90, 90])
Magnesium (hcp)#
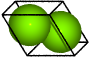
from ase.spacegroup import crystal
a = 3.21
c = 5.21
mg = crystal(
'Mg',
[(1.0 / 3.0, 2.0 / 3.0, 3.0 / 4.0)],
spacegroup=194,
cellpar=[a, a, c, 90, 90, 120],
)
Diamond#
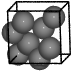
from ase.spacegroup import crystal
a = 3.57
diamond = crystal(
'C', [(0, 0, 0)], spacegroup=227, cellpar=[a, a, a, 90, 90, 90]
)
Sodium chloride#
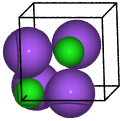
from ase.spacegroup import crystal
a = 5.64
nacl = crystal(
['Na', 'Cl'],
[(0, 0, 0), (0.5, 0.5, 0.5)],
spacegroup=225,
cellpar=[a, a, a, 90, 90, 90],
)
Rutile#
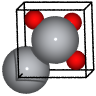
from ase.spacegroup import crystal
a = 4.6
c = 2.95
rutile = crystal(
['Ti', 'O'],
basis=[(0, 0, 0), (0.3, 0.3, 0.0)],
spacegroup=136,
cellpar=[a, a, c, 90, 90, 90],
)
CoSb3 skutterudite#
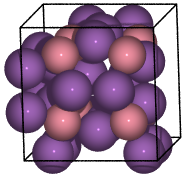
Skutterudites are quite interesting structures with 32 atoms in the unit cell.
from ase.spacegroup import crystal
a = 9.04
skutterudite = crystal(
('Co', 'Sb'),
basis=[(0.25, 0.25, 0.25), (0.0, 0.335, 0.158)],
spacegroup=204,
cellpar=[a, a, a, 90, 90, 90],
)
Often this structure is visualised with the Cobalt atoms on the
corners. This can easily be accomplished with ASE using
ase.build.cut(). Below is the origo argument used to
put the Cobalt atom on the corners and extend to include all corner
and edge atoms, even those belonging to neighbouring unit cells.
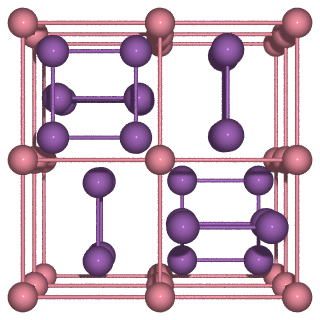
import ase.io as io
from ase.build import cut
from ase.spacegroup import crystal
a = 9.04
skutterudite = crystal(
('Co', 'Sb'),
basis=[(0.25, 0.25, 0.25), (0.0, 0.335, 0.158)],
spacegroup=204,
cellpar=[a, a, a, 90, 90, 90],
)
# Create a new atoms instance with Co at origo including all atoms on the
# surface of the unit cell
cosb3 = cut(skutterudite, origo=(0.25, 0.25, 0.25), extend=1.01)
# Define the atomic bonds to show
bondatoms = []
symbols = cosb3.get_chemical_symbols()
for i in range(len(cosb3)):
for j in range(i):
if symbols[i] == symbols[j] == 'Co' and cosb3.get_distance(i, j) < 4.53:
bondatoms.append((i, j))
elif (
symbols[i] == symbols[j] == 'Sb' and cosb3.get_distance(i, j) < 2.99
):
bondatoms.append((i, j))
# Create nice-looking image using povray
renderer = io.write(
'spacegroup-cosb3.pov',
cosb3,
rotation='90y',
radii=0.4,
povray_settings=dict(
transparent=False,
camera_type='perspective',
canvas_width=320,
bondlinewidth=0.07,
bondatoms=bondatoms,
),
)
renderer.render()
The Spacegroup class#
The ase.spacegroup.Spacegroup class is used
internally by the ase.spacegroup.crystal() function, but
might sometimes also be useful if you want to know e.g. the symmetry
operations of a given space group. Instances of the
ase.spacegroup.Spacegroup class are immutable
objects holding space group information, such as symmetry operations.
Let us e.g. consider the fcc structure. To print information about the space group, do
>>> from ase.spacegroup import Spacegroup
>>> sg = Spacegroup(225)
>>> print(sg)
225 F m -3 m
setting 1
centrosymmetric 1
primitive vectors
0.0000000000 0.5000000000 0.5000000000
0.5000000000 0.0000000000 0.5000000000
0.5000000000 0.5000000000 0.0000000000
reciprocal vectors
-1 1 1
1 -1 1
1 1 -1
4 subtranslations
0.0000000000 0.0000000000 0.0000000000
0.0000000000 0.5000000000 0.5000000000
0.5000000000 0.0000000000 0.5000000000
0.5000000000 0.5000000000 0.0000000000
24 symmetry operations (rot+trans)
1 0 0 0 1 0 0 0 1 0.0000000000 0.0000000000 0.0000000000
-1 0 0 0 -1 0 0 0 1 0.0000000000 0.0000000000 0.0000000000
-1 0 0 0 1 0 0 0 -1 0.0000000000 0.0000000000 0.0000000000
1 0 0 0 -1 0 0 0 -1 0.0000000000 0.0000000000 0.0000000000
0 0 1 1 0 0 0 1 0 0.0000000000 0.0000000000 0.0000000000
0 0 1 -1 0 0 0 -1 0 0.0000000000 0.0000000000 0.0000000000
0 0 -1 -1 0 0 0 1 0 0.0000000000 0.0000000000 0.0000000000
0 0 -1 1 0 0 0 -1 0 0.0000000000 0.0000000000 0.0000000000
0 1 0 0 0 1 1 0 0 0.0000000000 0.0000000000 0.0000000000
0 -1 0 0 0 1 -1 0 0 0.0000000000 0.0000000000 0.0000000000
0 1 0 0 0 -1 -1 0 0 0.0000000000 0.0000000000 0.0000000000
0 -1 0 0 0 -1 1 0 0 0.0000000000 0.0000000000 0.0000000000
0 1 0 1 0 0 0 0 -1 0.0000000000 0.0000000000 0.0000000000
0 -1 0 -1 0 0 0 0 -1 0.0000000000 0.0000000000 0.0000000000
0 1 0 -1 0 0 0 0 1 0.0000000000 0.0000000000 0.0000000000
0 -1 0 1 0 0 0 0 1 0.0000000000 0.0000000000 0.0000000000
1 0 0 0 0 1 0 -1 0 0.0000000000 0.0000000000 0.0000000000
-1 0 0 0 0 1 0 1 0 0.0000000000 0.0000000000 0.0000000000
-1 0 0 0 0 -1 0 -1 0 0.0000000000 0.0000000000 0.0000000000
1 0 0 0 0 -1 0 1 0 0.0000000000 0.0000000000 0.0000000000
0 0 1 0 1 0 -1 0 0 0.0000000000 0.0000000000 0.0000000000
0 0 1 0 -1 0 1 0 0 0.0000000000 0.0000000000 0.0000000000
0 0 -1 0 1 0 1 0 0 0.0000000000 0.0000000000 0.0000000000
0 0 -1 0 -1 0 -1 0 0 0.0000000000 0.0000000000 0.0000000000
Or, if you want to figure out what sites in the unit cell are equivalent to (0, 0, 0.5), simply do
>>> sites,kinds = sg.equivalent_sites([(0, 0, 0.5)])
>>> sites
array([[ 0. , 0. , 0.5],
[ 0.5, 0. , 0. ],
[ 0. , 0.5, 0. ],
[ 0.5, 0.5, 0.5]])
>>> kinds
[0, 0, 0, 0]
where sites will be an array containing the scaled positions of the four symmetry-equivalent sites.
- class ase.spacegroup.Spacegroup(spacegroup: int | str | Spacegroup, setting=1, datafile=None)[source]#
A space group class.
The instances of Spacegroup describes the symmetry operations for the given space group.
Example:
>>> from ase.spacegroup import Spacegroup >>> >>> sg = Spacegroup(225) >>> print('Space group', sg.no, sg.symbol) Space group 225 F m -3 m >>> sg.scaled_primitive_cell array([[ 0. , 0.5, 0.5], [ 0.5, 0. , 0.5], [ 0.5, 0.5, 0. ]]) >>> sites, kinds = sg.equivalent_sites([[0,0,0]]) >>> sites array([[ 0. , 0. , 0. ], [ 0. , 0.5, 0.5], [ 0.5, 0. , 0.5], [ 0.5, 0.5, 0. ]])
Returns a new Spacegroup instance.
Parameters:
- spacegroupint | string | Spacegroup instance
The space group number in International Tables of Crystallography or its Hermann-Mauguin symbol. E.g. spacegroup=225 and spacegroup=’F m -3 m’ are equivalent.
- setting1 | 2
Some space groups have more than one setting. \(setting\) determines Which of these should be used.
- datafileNone | string
Path to database file. If \(None\), the the default database will be used.
- ase.spacegroup.get_spacegroup(atoms, symprec=1e-05)[source]#
Determine the spacegroup to which belongs the Atoms object.
This requires spglib: https://atztogo.github.io/spglib/ .
Warning
The returned
Spacegroupobject has symmetry operations for a standard setting regardless of the givenAtomsobject. See ase/ase#1534 for details.Deprecated since version 3.24.0: Please use
ase.spacegroup.symmetrize.check_symmetryorspglibdirectly to get the symmetry operations for the givenAtomsobject.Parameters:
- atoms: Atoms object
Types, positions and unit-cell.
- symprec: float
Symmetry tolerance, i.e. distance tolerance in Cartesian coordinates to find crystal symmetry.
The Spacegroup object is returned.
Getting a reduced atomic basis#
You can also get a basis representation of a given crystal within a particular spagegroup,
using the ase.spacegroup.get_basis() function.
As an example, let’s look at rocksalt NaCl, and see how we can reproduce the basis from an ase.Atoms object:
>>> from ase.build import bulk
>>> from ase.spacegroup import get_basis
>>> atoms = bulk('NaCl', crystalstructure='rocksalt', a=5.64)
>>> spacegroup = 225 # Rocksalt
>>> basis = get_basis(atoms, spacegroup=spacegroup)
>>> basis
[[0. 0. 0. ]
[0.5 0.5 0.5]]
which gives us our expected 2 basis vectors for rocksalt, from the previous example.
Note
Inferring the spacegroup requires the installation of spglib, otherwise the space group must be passed explicitly.
- ase.spacegroup.get_basis(atoms: Atoms, spacegroup: int | str | Spacegroup = None, method: str = 'auto', tol: float = 1e-05) ndarray[source]#
Function for determining a reduced basis of an atoms object. Can use either an ASE native algorithm or an spglib based one. The native ASE version requires specifying a space group, while the (current) spglib version cannot. The default behavior is to automatically determine which implementation to use, based on the the
spacegroupparameter, and whether spglib is installed.- Parameters:
atoms – ase Atoms object to get basis from
spacegroup – Optional,
int,strorase.spacegroup.Spacegroupobject. If unspecified, the spacegroup can be inferred using spglib, if spglib is installed, andmethodis set to either'spglib'or'auto'. Inferring the spacegroup requires spglib.method –
str, one of:'auto'|'ase'|'spglib'. Selection of which implementation to use. It is recommended to use'auto', which is also the default.tol –
float, numeric tolerance for positional comparisons Default:1e-5