
Manipulating atoms#
Ag adatom on Ni slab#
We will set up a one layer slab of four Ni atoms with one Ag adatom. Define the slab atoms:
>>> from math import sqrt
>>> from ase import Atoms
>>> a = 3.55
>>> atoms = Atoms('Ni4',
... cell=[sqrt(2) * a, sqrt(2) * a, 1.0, 90, 90, 120],
... pbc=(1, 1, 0),
... scaled_positions=[(0, 0, 0),
... (0.5, 0, 0),
... (0, 0.5, 0),
... (0.5, 0.5, 0)])
>>> atoms.center(vacuum=5.0, axis=2)
Have a look at the cell and positions of the atoms:
>>> atoms.cell
Cell([[5.020458146424487, 0.0, 0.0], [-2.5102290732122423, 4.347844293440141, 0.0], [0.0, 0.0, 10.0]])
>>> atoms.positions
array([[ 0. , 0. , 5. ],
[ 2.51022907, 0. , 5. ],
[-1.25511454, 2.17392215, 5. ],
[ 1.25511454, 2.17392215, 5. ]])
>>> atoms[0]
Atom('Ni', [0.0, 0.0, 5.0], index=0)
Write the structure to a file and plot the whole system by bringing up the
ase.gui:
>>> from ase.visualize import view
>>> atoms.write('slab.xyz')
>>> view(atoms)
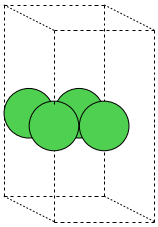
Within the viewer (called ase gui) it is possible to repeat
the unit cell in all three directions
(using the window).
From the command line, use ase gui -r 3,3,2 slab.xyz.
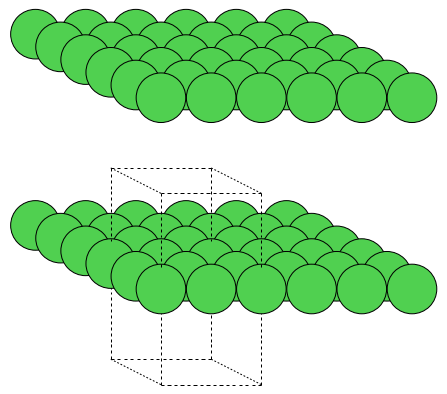
We now add an adatom in a three-fold site at a height of h=1.9 Å:
>>> h = 1.9
>>> relative = (1 / 6, 1 / 6, 0.5)
>>> absolute = np.dot(relative, atoms.cell) + (0, 0, h)
>>> atoms.append('Ag')
>>> atoms.positions[-1] = absolute
The structure now looks like this:
>>> view(atoms)
Interface building#
Now, we will make an interface with Ni(111) and water.
First we need a layer of water. One layer of water is constructed in this
script WL.py, and saved in the file WL.traj. Now run the
WL.py script and then read the atoms object from the traj file:
>>> from ase.io import read
>>> W = read('WL.traj')
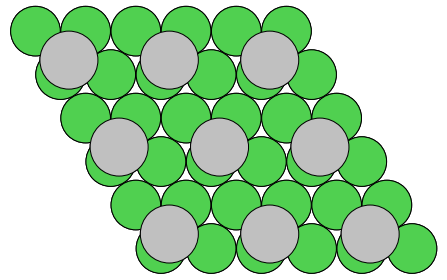
Lets take a look at the structure using view.
and let’s look at the unit cell.
>>> W.cell
Cell([8.490373, 4.901919, 26.93236])
We will need a Ni(111) slab which matches the water as closely as possible. A 2x4 orthogonal fcc111 supercell should be good enough.
>>> from ase.build import fcc111
>>> slab = fcc111('Ni', size=[2, 4, 3], a=3.55, orthogonal=True)

>>> slab.cell
Cell([5.020458146424487, 8.695688586880282, 0.0])
Looking at the two unit cells, we can see that they match with around 2 percent difference, if we rotate one of the cells 90 degrees in the plane. Let’s rotate the cell:
>>> W.cell = [W.cell[1, 1], W.cell[0, 0], 0.0]
Let’s also rotate() the molecules:
>>> W.rotate(90, 'z', center=(0, 0, 0))
Now we can wrap the atoms into the cell
>>> W.wrap()
The wrap() method only works if periodic boundary
conditions are enabled. We have a 2 percent lattice mismatch between Ni(111)
and the water, so we scale the water in the plane to match the cell of the
slab.
The argument scale_atoms=True indicates that the atomic positions should be
scaled with the unit cell. The default is scale_atoms=False indicating that
the cartesian coordinates remain the same when the cell is changed.
>>> W.set_cell(slab.cell, scale_atoms=True)
>>> zmin = W.positions[:, 2].min()
>>> zmax = slab.positions[:, 2].max()
>>> W.positions += (0, 0, zmax - zmin + 1.5)
Finally we use extend to copy the water onto the slab:
>>> interface = slab + W
>>> interface.center(vacuum=6, axis=2)
>>> interface.write('NiH2O.traj')
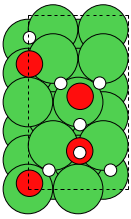
Adding two atoms objects will take the positions from both and the cell and boundary conditions from the first.