Linear dielectric response of an extended system¶
A short introduction¶
The DielectricFunction object can calculate the dielectric function of an extended system from its ground state electronic structure. The frequency and wave-vector dependent linear dielectric matrix in reciprocal space representation is written as
where \(\mathbf{q}\) and \(\omega\) are the momentum and energy transfer in an excitation, and \(\mathbf{G}\) is reciprocal lattice vector. The off-diagonal element of \(\epsilon_{\mathbf{G} \mathbf{G}^{\prime}}\) determines the local field effect.
The macroscopic dielectric function (with local field correction) is defined by
Ignoring the local field (the off-diagonal element of dielectric matrix) results in:
Optical absorption spectrum is obtained through
Electron energy loss spectrum (EELS) is obtained by
The macroscopic dielectric function is ill defined for systems of reduced dimensionality. In these cases \(\epsilon_M = 1.0\). The polarizability will maintain its structure for lower dimensionalities and is in 3D related to the macroscopic dielectric function as,
Refer to Linear dielectric response of an extended system: theory for detailed documentation on theoretical part.
Frequency grid¶
The dielectric function is evaluated on a non-linear frequency grid according to the formula
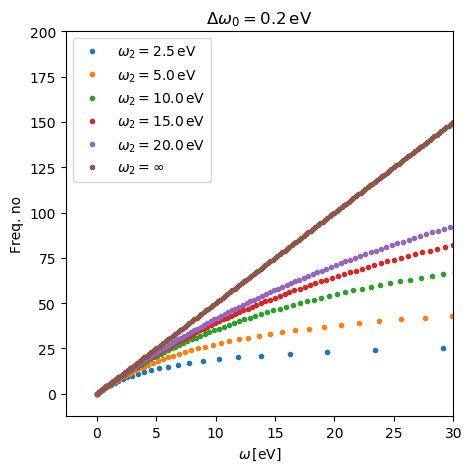
The parameter \(\Delta\omega_0\) is the frequency spacing at \(\omega=0\) and \(\omega_2\) is the frequency where the spacing has increased to \(2\Delta\omega_0\). In general a lower value of \(\omega_2\) gives a more non- linear grid and less frequency points.
Below, the frequency grid is visualized for different values of
\(\omega_2\). You can find the script for reproducing this figure here:
plot_freq.py.
The parameters can be specified using keyword arguments:
df = DielectricFunction(
...,
frequencies={'type': 'nonlinear', # frequency grid specification
'domega0: 0.05, # eV. Default = 0.1 eV
'omega2': 5.0, # eV. Default = 10.0 eV
'omegamax': 15.0}) # eV. Default is the maximum
# difference between energy
# eigenvalues
Setting omegamax manually is usually not advisable, however you
might want it in cases where semi-core states are included where very large
energy eigenvalue differences appear.
- class gpaw.response.frequencies.FrequencyDescriptor(omega_w)[source]¶
Frequency grid descriptor.
- Parameters:
omega_w (ArrayLike1D) – Frequency grid in Hartree units.
- static from_array_or_dict(input)[source]¶
Create frequency-grid descriptor.
In case input is a list on frequencies (in eV) a
FrequencyGridDescriptorinstance is returned. Othervise aNonLinearFrequencyDescriptorinstance is returned.>>> from ase.units import Ha >>> params = dict(type='nonlinear', ... domega0=0.1, ... omega2=10, ... omegamax=50) >>> wd = FrequencyDescriptor.from_array_or_dict(params) >>> wd.omega_w[0:2] * Ha array([0. , 0.10041594])
- class gpaw.response.frequencies.NonLinearFrequencyDescriptor(domega0, omega2, omegamax)[source]¶
Non-linear frequency grid.
Units are Hartree. See Frequency grid.
- Parameters:
Example 1: Optical absorption of semiconductor: Bulk silicon¶
A simple startup¶
Here is a minimum script to get an absorption spectrum.
from ase.build import bulk
from gpaw import GPAW
from gpaw.response.df import DielectricFunction
# Part 1: Ground state calculation
atoms = bulk('Si', 'diamond', a=5.431)
calc = GPAW(mode='pw', kpts=(4, 4, 4))
atoms.calc = calc
atoms.get_potential_energy() # ground state calculation is performed
calc.write('si.gpw', 'all') # use 'all' option to write wavefunction
# Part 2 : Spectrum calculation
# DF: dielectric function object
# Ground state gpw file (with wavefunction) as input
df = DielectricFunction(
calc='si.gpw',
frequencies={'type': 'nonlinear',
'domega0': 0.05}, # using nonlinear frequency grid
rate='eta')
# By default, a file called 'df.csv' is generated
df.get_dielectric_function()
This script takes less than one minute on a single cpu, and generates a file ‘df.csv’ containing the optical (\(\mathbf{q} = 0\)) dielectric function along the x-direction, which is the default direction. The file ‘df.csv’ contain five columns ordered as follows: \(\omega\) (eV), \(\mathrm{Re}(\epsilon_{\mathrm{NLF}})\), \(\mathrm{Im}(\epsilon_{\mathrm{NLF}})\), \(\mathrm{Re}(\epsilon_{\mathrm{LF}})\), \(\mathrm{Im}(\epsilon_{\mathrm{LF}})\) where \(\epsilon_{\mathrm{NLF}}\) and \(\epsilon_{\mathrm{LF}}\) is the result without and with local field effects, respectively.
For other directions you can specify the direction and filename like:
DielectricFunction.get_dielectric_function(...,
direction='y',
filename='filename.csv',
...)
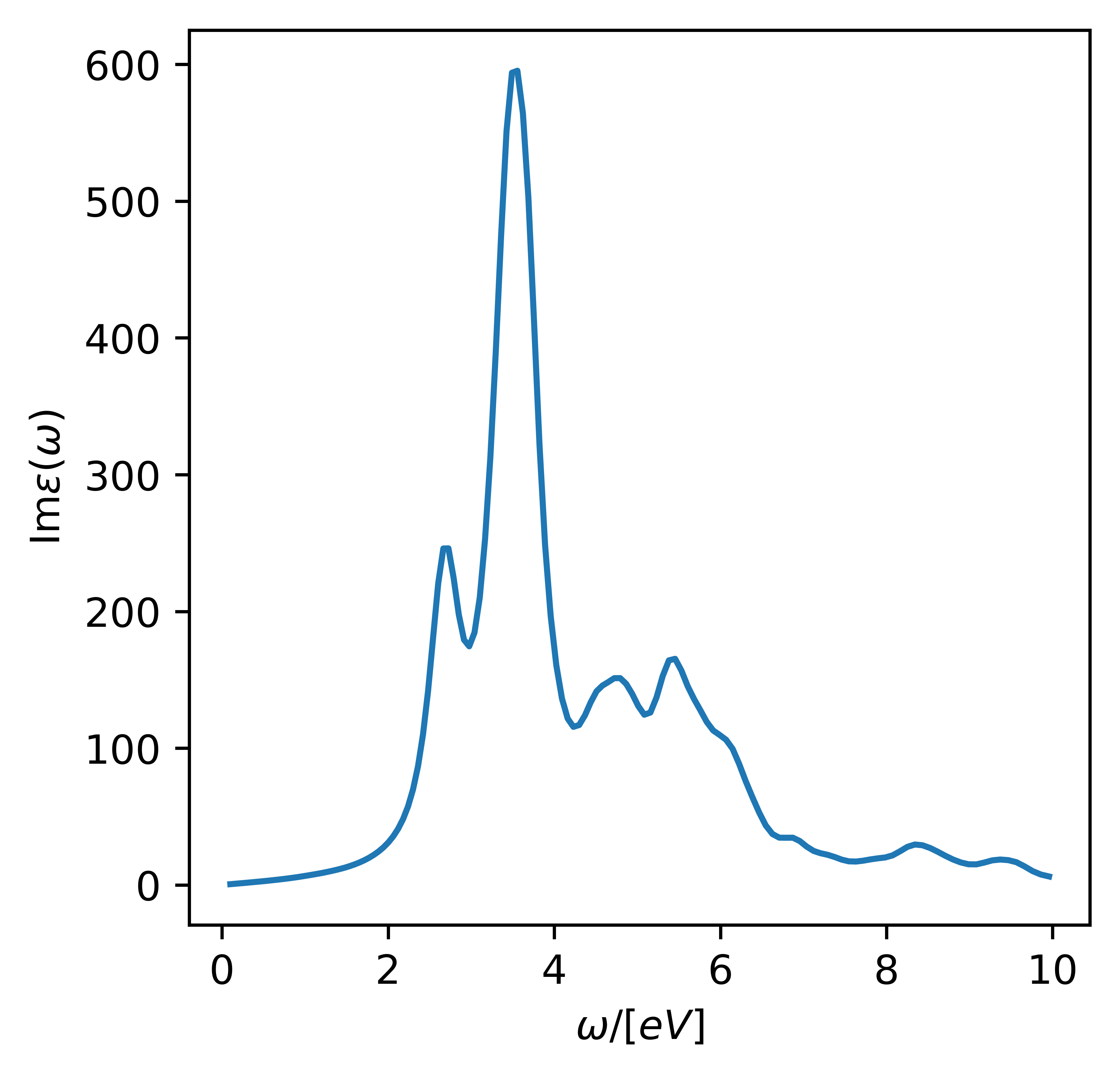
The absorption spectrum along the x-direction including local field effects can then be plotted using
# web-page: si_abs.png
import numpy as np
from math import pi
import matplotlib.pyplot as plt
from matplotlib import rc
rc('font', **{'family': 'sans-serif', 'sans-serif': ['Helvetica']})
# rc('text', usetex=True)
rc('figure', figsize=(4.0, 4.0), dpi=800)
data = np.loadtxt('df.csv', delimiter=',')
# Set plotting range
xmin = 0.1
xmax = 10.0
inds_w = (data[:, 0] >= xmin) & (data[:, 0] <= xmax)
plt.plot(data[inds_w, 0], 4 * pi * data[inds_w, 4])
plt.xlabel('$\\omega / [eV]$')
plt.ylabel('$\\mathrm{Im} \\epsilon(\\omega)$')
plt.savefig('si_abs.png', bbox_inches='tight')
The resulting figure is shown below. Note that there is significant absorption close to \(\omega=0\) because of the large default Fermi-smearing in GPAW. This will be dealt with in the following more realistic calculation.
More realistic calculation¶
To get a realistic silicon absorption spectrum and macroscopic dielectric
constant, one needs to converge the calculations with respect to grid
spacing, kpoints, number of bands, planewave cutoff energy and so on. Here
is an example script: silicon_ABS.py. In the following, the script
is split into different parts for illustration.
Ground state calculation
# Refer to G. Kresse, Phys. Rev. B 73, 045112 (2006) # for comparison of macroscopic and microscopic dielectric constant # and absorption peaks. from pathlib import Path from ase.build import bulk from ase.parallel import paropen, world from gpaw import GPAW, FermiDirac from gpaw.response.df import DielectricFunction # Ground state calculation a = 5.431 atoms = bulk('Si', 'diamond', a=a) calc = GPAW(mode='pw', kpts={'density': 5.0, 'gamma': True}, parallel={'band': 1, 'domain': 1}, xc='LDA', occupations=FermiDirac(0.001)) # use small FD smearing atoms.calc = calc atoms.get_potential_energy() # get ground state density # Restart Calculation with fixed density and dense kpoint sampling calc = calc.fixed_density( kpts={'density': 15.0, 'gamma': False}) # dense kpoint sampling calc.diagonalize_full_hamiltonian(nbands=70) # diagonalize Hamiltonian calc.write('si_large.gpw', 'all') # write wavefunctionsIn this script a normal ground state calculation is performed with coarse kpoint grid. The calculation is then restarted with a fixed density and the full hamiltonian is diagonalized exactly on a densely sampled kpoint grid. This is the preferred procedure of obtained the excited KS-states because it is in general difficult to converge the excited states using iterative solvers. A full diagonalization is more robust.
Note
For semiconductors, it is better to use either small Fermi-smearing in the ground state calculation:
from gpaw import FermiDirac calc = GPAW(... occupations=FermiDirac(0.001), ...)or larger ftol, which determines the threshold for transition in the dielectric function calculation (\(f_i - f_j > ftol\)), not shown in the example script):
df = DielectricFunction(... ftol=1e-2, ...)
Get absorption spectrum
# Getting absorption spectrum df = DielectricFunction(calc='si_large.gpw', eta=0.05, frequencies={'type': 'nonlinear', 'domega0': 0.02}, ecut=150) df.get_dielectric_function(filename='si_abs.csv')Here
etais the broadening parameter of the calculation, andecutis the local field effect cutoff included in the dielectric function.
Get macroscopic dielectric constant
The macroscopic dielectric constant is defined as the real part of dielectric function at \(\omega=0\). In the following script, only a single point at \(\omega=0\) is calculated without using the hilbert transform (which is only compatible with the non-linear frequency grid specification).
# Getting macroscopic constant df = DielectricFunction(calc='si_large.gpw', frequencies=[0.0], hilbert=False, eta=0.0001, ecut=150, ) epsNLF, epsLF = df.get_macroscopic_dielectric_constant() # Make table epsrefNLF = 14.08 # from [1] in top epsrefLF = 12.66 # from [1] in top with paropen('mac_eps.csv', 'w') as f: print(' , Without LFE, With LFE', file=f) print(f"{'GPAW-linear response'}, {epsNLF:.6f}, {epsLF:.6f}", file=f) print(f"{'[1]'}, {epsrefNLF:.6f}, {epsrefLF:.6f}", file=f) print(f"{'Exp.'}, {11.9:.6f}, {11.9:.6f}", file=f) if world.rank == 0: Path('si_large.gpw').unlink()In general, local field correction will reduce this value by 10-20%.
Result¶
The figure shown here is generated from script :
silicon_ABS.py and plot_ABS.py.
It takes 30 minutes with 16 cpus on Intel Xeon X5570 2.93GHz.
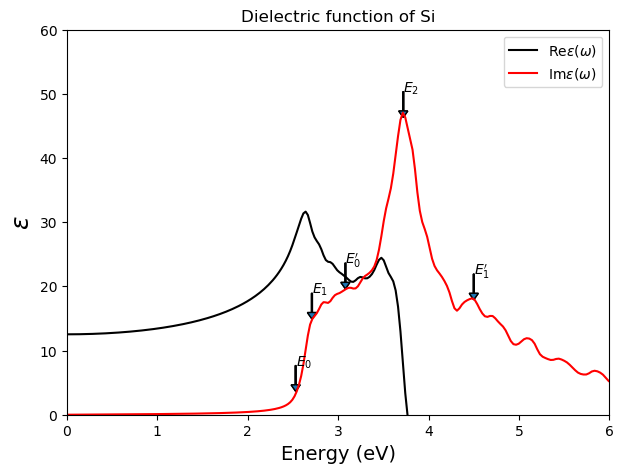
The arrows are data extracted from [1].
The calculated macroscopic dielectric constant can be seen in the table below and compare good with the values from [1]. The experimental value is 11.90. The larger theoretical value results from the fact that the ground state LDA (even GGA) calculation underestimates the bandgap.
Without LFE |
With LFE |
|
GPAW-linear response |
13.935579 |
12.533488 |
[1] |
14.080000 |
12.660000 |
Exp. |
11.900000 |
11.900000 |
Example 2: Electron energy loss spectra¶
Electron energy loss spectra (EELS) can be used to explore the plasmonic (collective electronic) excitations of an extended system. This is because the energy loss of a fast electron passing by a material is defined by
and the plasmon frequency \(\omega_p\) is defined as \(\epsilon(\omega_p) \rightarrow 0\). It means that an external perturbation at this frequency, even infinitesimal, can generate a large collective electronic response.
A simple startup: bulk aluminum¶
Here is a minimum script to get an EELS spectrum.
from ase.build import bulk
from gpaw import GPAW
from gpaw.response.df import DielectricFunction
# Part 1: Ground state calculation
atoms = bulk('Al', 'fcc', a=4.043) # generate fcc crystal structure
# GPAW calculator initialization:
k = 13
calc = GPAW(mode='pw', kpts=(k, k, k))
atoms.calc = calc
atoms.get_potential_energy() # ground state calculation is performed
calc.write('Al.gpw', 'all') # use 'all' option to write wavefunctions
# Part 2: Spectrum calculation
df = DielectricFunction(calc='Al.gpw') # ground state gpw file as input
# Momentum transfer, must be the difference between two kpoints:
q_c = [1.0 / k, 0, 0]
df.get_eels_spectrum(q_c=q_c) # by default, an 'eels.csv' file is generated
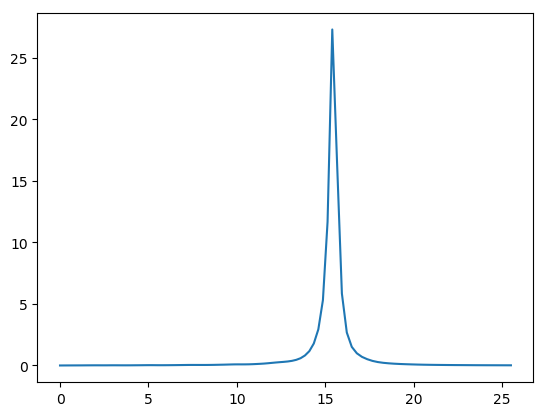
This script takes less than one minute on a single cpu and by default, generates a file ‘EELS.csv’. Then you can plot the file using
# web-page: aluminum_EELS.png
import numpy as np
import matplotlib.pyplot as plt
data = np.loadtxt('eels.csv', delimiter=',')
plt.plot(data[:, 0], data[:, 2])
plt.savefig('aluminum_EELS.png', bbox_inches='tight')
plt.show()
The three columns of this file correspond to energy (eV), EELS without and with local field correction, respectively. You will see a 15.9 eV peak. It comes from the bulk plasmon excitation of aluminum. You can explore the plasmon dispersion relation \(\omega_p(\mathbf{q})\) by tuning \(\mathbf{q}\) in the calculation above.
Note
The momentum transfer \(\mathbf{q}\) in an EELS calculation must be the difference between two kpoints! For example, if you have an kpts=(Nk1, Nk2, Nk3) Monkhorst-Pack k-sampling in the ground state calculation, you have to choose \(\mathbf{q} = \mathrm{np.array}([i/Nk1, j/Nk2, k/Nk3])\), where \(i, j, k\) are integers.
A more sophisticated example: graphite¶
Here is a more sophisticated example of calculating EELS of graphite with different \(\mathbf{q}\). You can also get the script here: graphite_EELS.py. The results (plot) are shown in the following section.
from math import sqrt
import numpy as np
from ase import Atoms
from ase.parallel import paropen
from gpaw import GPAW, FermiDirac, PW
from gpaw.response.df import DielectricFunction
# Part 1: Ground state calculation
a = 1.42
c = 3.355
# Generate graphite AB-stack structure:
atoms = Atoms('C4',
scaled_positions=[(1 / 3.0, 1 / 3.0, 0),
(2 / 3.0, 2 / 3.0, 0),
(0, 0, 0.5),
(1 / 3.0, 1 / 3.0, 0.5)],
pbc=(1, 1, 1),
cell=[(sqrt(3) * a / 2, 3 / 2.0 * a, 0),
(-sqrt(3) * a / 2, 3 / 2.0 * a, 0),
(0, 0, 2 * c)])
# Part 2: Find ground state density and diagonalize full hamiltonian
calc = GPAW(mode=PW(500),
kpts=(6, 6, 3),
parallel={'domain': 1},
# Use smaller Fermi-Dirac smearing to avoid intraband transitions:
occupations=FermiDirac(0.05))
atoms.calc = calc
atoms.get_potential_energy()
calc = calc.fixed_density(kpts=(20, 20, 7))
# The result should also be converged with respect to bands:
calc.diagonalize_full_hamiltonian(nbands=60)
calc.write('graphite.gpw', 'all')
# Part 2: Spectra calculations
f = paropen('graphite_q_list', 'w') # write q
for i in range(1, 6): # loop over different q
df = DielectricFunction(calc='graphite.gpw',
frequencies={'type': 'nonlinear',
'domega0': 0.01},
eta=0.2, # Broadening parameter.
ecut=100,
# write different output for different q:
txt='out_df_%d.txt' % i)
q_c = [i / 20.0, 0.0, 0.0] # Gamma - M excitation
df.get_eels_spectrum(q_c=q_c, filename='graphite_EELS_%d' % i)
# Calculate cartesian momentum vector:
cell_cv = atoms.get_cell()
bcell_cv = 2 * np.pi * np.linalg.inv(cell_cv).T
q_v = np.dot(q_c, bcell_cv)
print(sqrt(np.inner(q_v, q_v)), file=f)
f.close()
Results on graphite¶
The figure shown here is generated from script: graphite_EELS.py
and plot_EELS.py
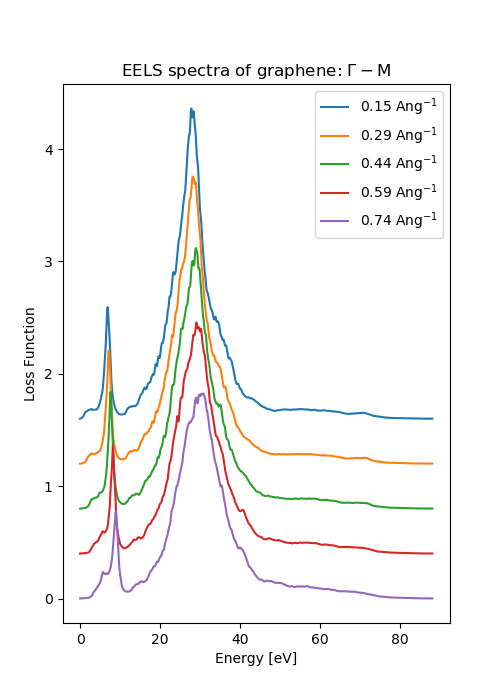
One can compare the results with literature [2].
Example 3: Tetrahedron integration (experimental)¶
The k-point integral needed for the calculation of the density response function, is typically performed using a simple summation of individual k-points. By setting the integration mode to ‘tetrahedron integration’ the kpoint integral is calculated by interpolating density matrix elements and eigenvalues. This typically means that fewer k-points are needed for convergence.
The combination of using the tetrahedron method and reducing the kpoint integral using crystal symmetries means that it is necessary to be careful with the kpoint sampling which should span the whole irreducible zone. Luckily, bztools.py provides the tool to generate such an k-point sampling autimatically:
from gpaw.bztools import find_high_symmetry_monkhorst_pack
kpts = find_high_symmetry_monkhorst_pack(
'gs.gpw', # Path to ground state .gpw file
density=20.0) # The required minimum density
Bulk TaS2¶
As an example, lets calculate the optical properties of a well-known layered
material, namely, the transition metal dichalcogenide (TMD) 2H-TaS2. We set the structure and find the ground state density with (find the full
script here tas2_dielectric_function.py, takes about 10 mins on 8
cores.)
# web-page: tas2_eps.png
from ase import Atoms
from ase.lattice.hexagonal import Hexagonal
import matplotlib.pyplot as plt
from gpaw import GPAW, PW, FermiDirac
from gpaw.response.df import DielectricFunction
from gpaw.mpi import world
from gpaw.bztools import find_high_symmetry_monkhorst_pack
# 1) Ground-state.
a = 3.314
c = 12.1
cell = Hexagonal(symbol='Ta', latticeconstant={'a': a, 'c': c}).get_cell()
atoms = Atoms(symbols='TaS2TaS2', cell=cell, pbc=(1, 1, 1),
scaled_positions=[(0, 0, 1 / 4),
(2 / 3, 1 / 3, 1 / 8),
(2 / 3, 1 / 3, 3 / 8),
(0, 0, 3 / 4),
(-2 / 3, -1 / 3, 5 / 8),
(-2 / 3, -1 / 3, 7 / 8)])
calc = GPAW(mode=PW(600),
xc='PBE',
occupations=FermiDirac(width=0.01),
kpts={'density': 5})
atoms.calc = calc
atoms.get_potential_energy()
calc.write('TaS2-gs.gpw')
The next step is to calculate the unoccupied bands
kpts = find_high_symmetry_monkhorst_pack('TaS2-gs.gpw', density=5.0)
responseGS = GPAW('TaS2-gs.gpw').fixed_density(
kpts=kpts,
parallel={'band': 1},
nbands=60,
convergence={'bands': 50})
responseGS.get_potential_energy()
responseGS.write('TaS2-gsresponse.gpw', 'all')
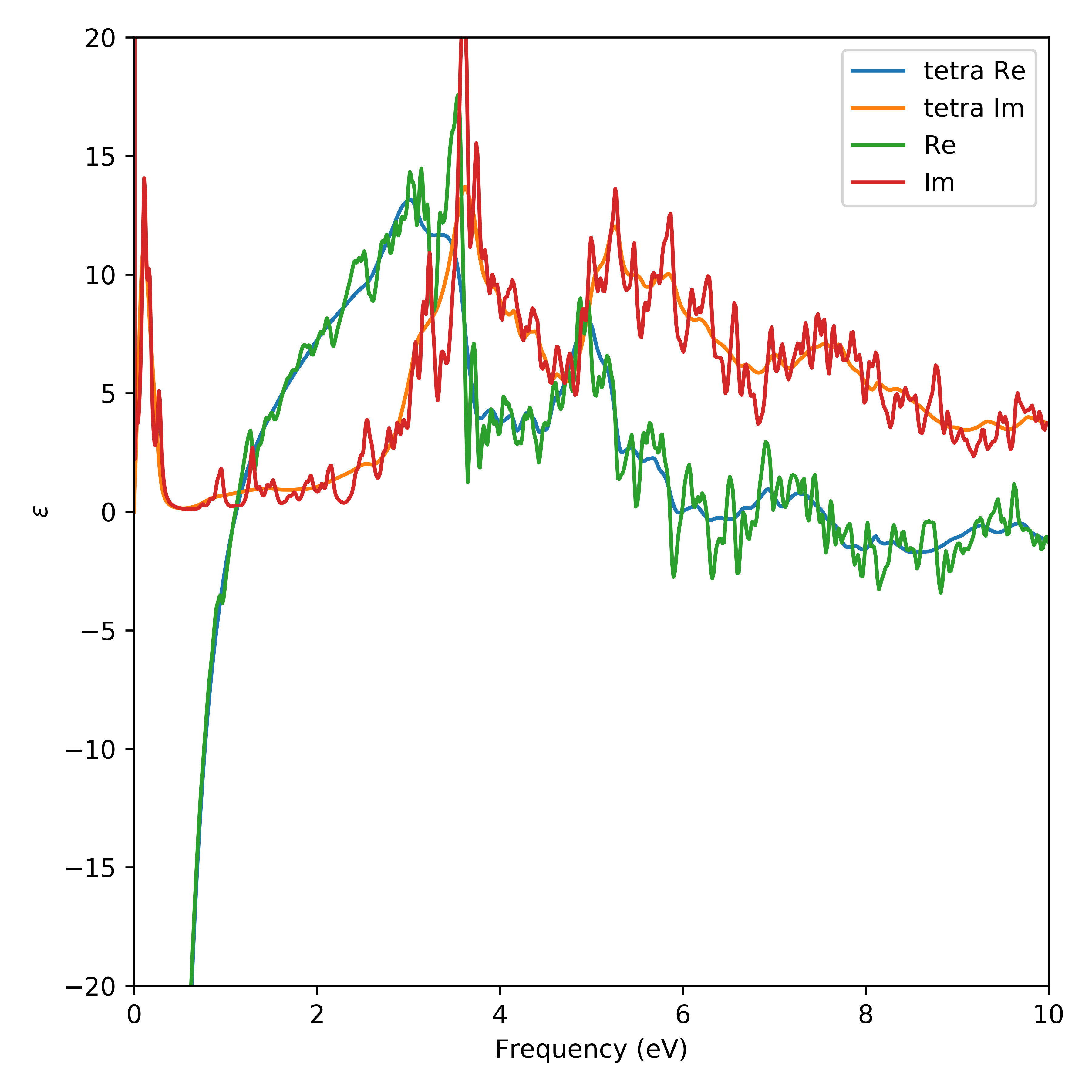
Lets compare the result of the tetrahedron method with the point sampling
method for broadening of eta=25 meV.
df = DielectricFunction(
'TaS2-gsresponse.gpw',
eta=25e-3,
rate='eta',
frequencies={'type': 'nonlinear', 'domega0': 0.01},
integrationmode='tetrahedron integration')
df1tetra_w, df2tetra_w = df.get_dielectric_function(direction='x')
df = DielectricFunction(
'TaS2-gsresponse.gpw',
eta=25e-3,
rate='eta',
frequencies={'type': 'nonlinear', 'domega0': 0.01})
df1_w, df2_w = df.get_dielectric_function(direction='x')
omega_w = df.get_frequencies()
if world.rank == 0:
plt.figure(figsize=(6, 6))
plt.plot(omega_w, df2tetra_w.real, label='tetra Re')
plt.plot(omega_w, df2tetra_w.imag, label='tetra Im')
plt.plot(omega_w, df2_w.real, label='Re')
plt.plot(omega_w, df2_w.imag, label='Im')
plt.xlabel('Frequency (eV)')
plt.ylabel('$\\varepsilon$')
plt.xlim(0, 10)
plt.ylim(-20, 20)
plt.legend()
plt.tight_layout()
plt.savefig('tas2_eps.png', dpi=600)
# plt.show()
The result of which is shown below. Comparing the methods shows that the calculated dielectric function is much more well behaved when calculated using the tetrahedron method.
Graphene¶
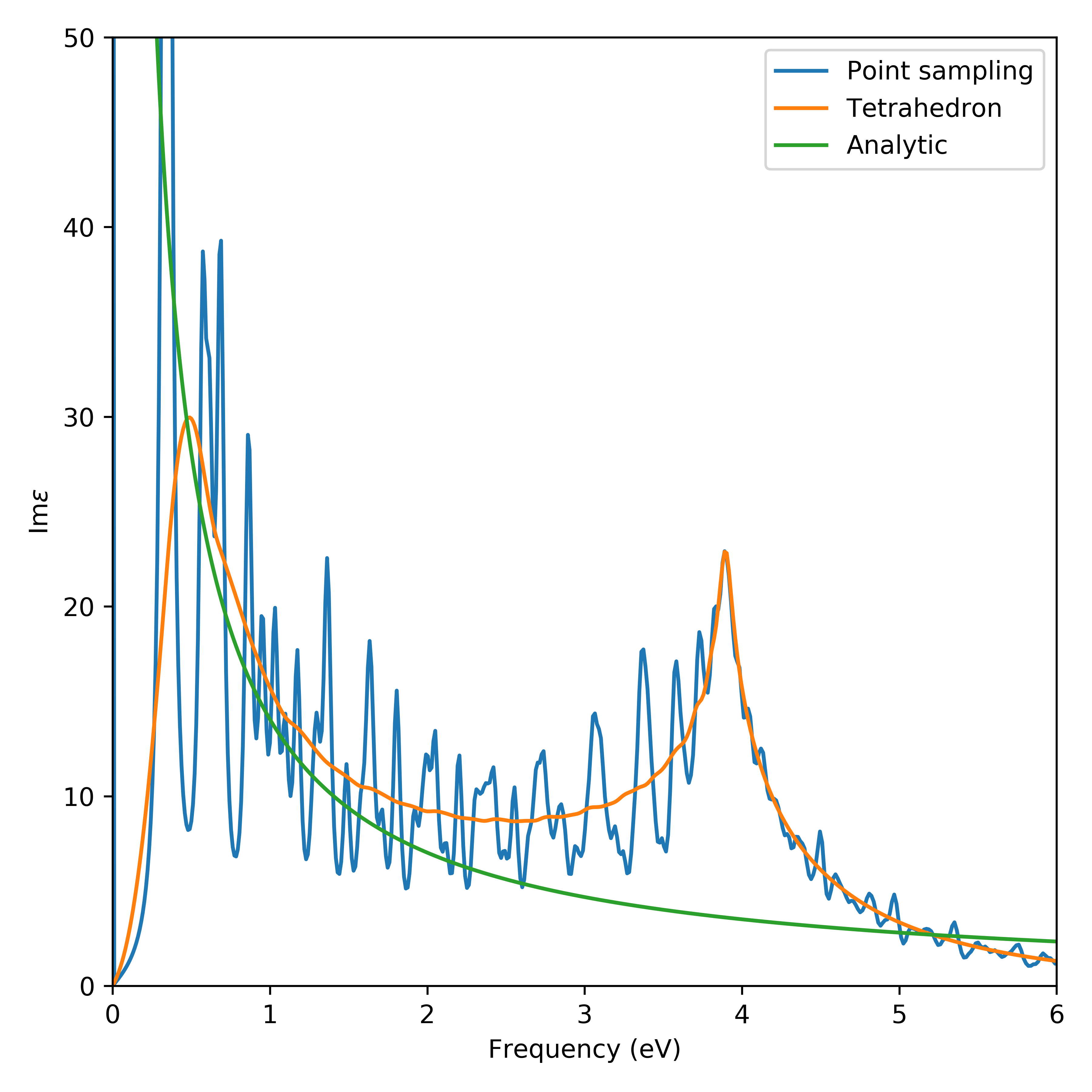
Graphene represents a material which is particularly difficult to converge with respect to the number of k-points. This property originates from its semi-metallic nature and vanishing density of states at the fermi-level which make the calculated properties sensitive to the k-point distribution in the vicinity of the Dirac point. Since the macroscopic dielectric function is not well-defined for two-dimensional systems we will focus on calculating the dielectric function an artificial structure of graphite with double the out-of-plane lattice constant. This should make its properties similar to graphene. It is well known that graphene has a sheet conductance of \(\sigma_s=\frac{e^2}{4\hbar}\) so, if the individual graphite layers behave as ideal graphene, the optical properties of the structure should be determined by
where \(d=3.22\) Å is the thickness of graphene. Below we show the
calculated dielectic function
(graphene_dielectric_function.py) for a minimum
k-point density of 30 Å\(^{-1}\) corresponding to a kpoint sampling of size
(90, 90, 1) and room temperatur broadening eta = 25 meV. It
is clear that the properties are well converged for large frequencies
using the tetrahedron method. For smaller frequencies the tetrahedron
method fares comparably but convergence is tough for frequencies below
0.5 eV where the absorption spectrum goes to zero. This behaviour is
due to the partially occupied Dirac point which sensitively effects the
integrand of the density response function.
k-point convergence comparison¶
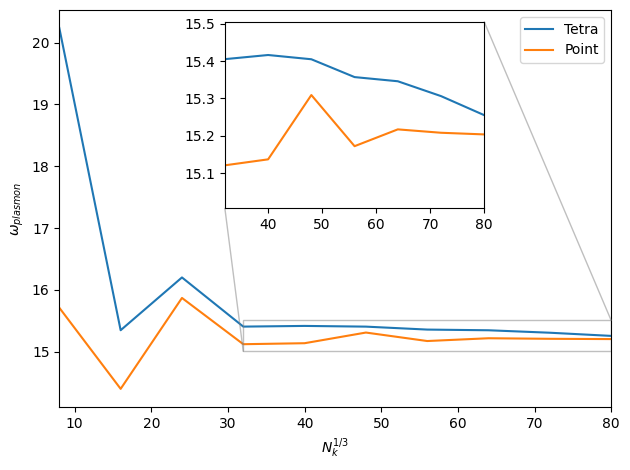
Elemental aluminium is another material which can be difficult to
converge with respect to the number of k-points.
This is due to the Fermi surface of the metal penetrating
the surface of the first Brilluoin zone.
This means that the fermi surface has to be finely resolved when
calculating ´q = 0´ EELS spectra.
The EELS spectrum of Al shows a clear plasmonic resonsance and
below we show the k-point convergence of the plasmon frequency.
We compare the tetrahedron integration described here with the
default point integration method.
The dielectric function is calculated for all varying k-samplings
(al-plasmon-peak.py) using ´Gamma´-centered Monkhorst-Pack
k-point grids with a multiple of 8 points along each axis to ensure sampling of
high-symmetry k-points. We see that both methods converge to the same value.
Notes on the implementation of the tetrahedron method¶
The tetrahedron method is still in an experimental phase which means that the user should check the results against the results obtained with point integration. The method is based on the paper of [3] whom provide analytical expressions for the response function integration with interpolated eigenvalues and matrix elements.
In other implementations of the tetrahedron integration method, the division
of the BZ into tetrahedras has been performed using a recursive scheme. In
our implementation we use the external library scipy.spatial which
provide the Delaunay() class to calculate Delaunay triangulated k-point
grids from the ground state k-point grid. Delaunay triangulated grids do
necessarily give a good division of the Brillouin zone into tetrahedras and
particularly for homogeneously spaced k-point grids (such as a Monkhorst
Pack) grid many ill-defined tetrahedrons may be created. Such tetrahedrons
are however easily filtered away by removing tetrahedrons with very small
volumes.
Suggested performance enhancement:
TetrahedronDescriptor(kpts, simplices=None)Factor out the creation of tetrahedras to make it possible to use user-defined tetrahedras. Such an object could inherit from theDelaunay()class.
Additionally, this library provides a k-dimensional tree implementation
cKDTree() which is used to efficiently identify k-points in the Brillouin
zone. For finite \(\mathbf{q}\) it is still necessary for the kpoint grid to be
uniform. In the optical limit where \(\mathbf{q}=0\), however, it is possible
to use non-uniform k-point grids which can aid a faster convergence,
especially for materials such as graphene.
When using the tetrahedron integration method, the plasmafrequency is calculated at \(T=0\) where the expression reduces an integral over the Fermi surface. The interband transitions are calculated using the temperature of the ground state calculation. The occupation factors are included as modifications to the matrix elements as \(\sqrt{f_n - f_m} \langle \psi_m|\nabla |\psi_n \rangle\)
Suggested performance enhancement: Take advantage of the analytical dependence of the occupation numbers to treat partially occupied bands more accurately. This is expected to improve the convergence of the optical properties of graphene for low frequencies.
The entirety of the tetrahedron integration code is written in Python except for the calculation of the algebraic expression for the response function integration of a single tetrahedron for which the Python overhead is significant. The algebraic expression are therefore evaluated in C.
Technical details:¶
There are few points about the implementation that we emphasize:
The code is parallelized over kpoints and occupied bands. The parallelization over occupied bands makes it straight-forward to utilize efficient BLAS libraries to sum un-occupied bands.
The code employs the Hilbert transform in which the spectral function for the density-density response function is calculated before calculating the full density response. This speeds up the code significantly for calculations with a lot of frequencies.
The non-linear frequency grid employed in the calculations is motivated by the fact that when using the Hilbert transform the real part of the dielectric function converges slowly with the upper bound of the frequency grid. Refer to Linear dielectric response of an extended system: theory for the details on the Hilbert transform.
Drude phenomenological scattering rate¶
A phenomenological scattering rate can be introduced using the rate
parameter in the optical limit. By default this is zero but can be set by
using:
df = DielectricFunction(...
rate=0.1,
...)
to set a scattering rate of 0.1 eV. If rate=’eta’ then the code with use the
specified eta parameter. Note that the implementation of the rate parameter
differs from some literature by a factor of 2 for consistency with the linear
response formalism. In practice the Drude rate is implemented as
where \(\gamma\) is the rate parameter.
Useful tips¶
Use --dry-run option to get an overview of a calculation
(especially useful for heavy calculations!):
$ gpaw python --dry-run=8 filename.py
Note
But be careful ! LCAO mode calculation results in unreliable unoccupied states above vacuum energy.
It’s important to converge the results with respect to:
nbands,
nkpt (number of kpoints in gs calc.),
eta,
ecut,
ftol,
omegamax (the maximum energy, be careful if hilbert transform is used)
domega0 (the energy spacing, if there is)
vacuum (if there is)
Parameters¶
keyword |
type |
default value |
description |
|---|---|---|---|
|
|
None |
gpw filename (with ‘all’ option when writing the gpw file) |
|
|
None |
If specified the chi matrix is
saved to |
|
|
None |
Energies for spectrum. If left unspecified the frequency grid will be non-linear. Ex: numpy.linspace(0,20,201) |
|
|
0.1 |
\(\Delta\omega_0\) for non-linear frequency grid. |
|
|
10.0 (eV) |
\(\omega_2\) for non-linear frequencygrid. |
|
|
Maximum energy eigenvalue difference. |
Maximum frequency. |
|
|
10 (eV) |
Planewave energy cutoff. Determines the size of dielectric matrix. |
|
|
0.2 (eV) |
Broadening parameter. |
|
|
1e-6 |
The threshold for transition: \(f_{ik} - f_{jk} > ftol\) |
|
|
stdout |
Output filename. |
|
|
True |
Switch for hilbert transform. |
|
|
nbands from gs calc |
Number of bands from gs calc to include. |
|
|
0.0 (eV) |
Phenomenological Drude scattering rate. If rate=”eta” then use “eta”. Note that this may differ by a factor of 2 for some definitions of the Drude scattering rate. See the section on Drude scattering rate. |
Details of the DF object¶
- class gpaw.response.df.DielectricFunction(calc, *, frequencies=None, ecut=50, hilbert=True, nbands=None, eta=0.2, intraband=True, nblocks=1, world=MPIComm(size=1, rank=0), txt=<_io.TextIOWrapper name='<stdout>' mode='w' encoding='utf-8'>, truncation=None, qsymmetry=True, integrationmode=None, rate=0.0, eshift=None)[source]¶
This class defines dielectric function related physical quantities.
Creates a DielectricFunction object.
- calc: str
The ground-state calculation file that the linear response calculation is based on.
- frequencies:
Input parameters for frequency_grid. Can be an array of frequencies to evaluate the response function at or dictionary of parameters for build-in nonlinear grid (see Frequency grid).
- ecut: float
Plane-wave cut-off.
- hilbert: bool
Use hilbert transform.
- nbands: int
Number of bands from calculation.
- eta: float
Broadening parameter.
- intraband: bool
Include intraband transitions.
- world: comm
mpi communicator.
- nblocks: int
Split matrices in nblocks blocks and distribute them G-vectors or frequencies over processes.
- txt: str
Output file.
- truncation: str or None
None for no truncation. ‘2D’ for standard analytical truncation scheme. Non-periodic directions are determined from k-point grid
- eshift: float
Shift unoccupied bands
- get_dielectric_function(*args, filename='df.csv', **kwargs)[source]¶
Calculate the dielectric function.
Generates a file ‘df.csv’, unless filename is set to None.
- Returns:
df_NLFC_w (np.ndarray) – Dielectric function without local field corrections.
df_LFC_w (np.ndarray) – Dielectric functio with local field corrections.
- get_eels_spectrum(*args, filename='eels.csv', **kwargs)[source]¶
Calculate the macroscopic EELS spectrum.
Generates a file ‘eels.csv’, unless filename is set to None.
- Returns:
eels0_w (np.ndarray) – Spectrum in the independent-particle random-phase approximation.
eels_w (np.ndarray) – Fully screened EELS spectrum.
- get_macroscopic_dielectric_constant(xc='RPA', direction='x')[source]¶
Calculate the macroscopic dielectric constant.
The macroscopic dielectric constant is defined as the real part of the dielectric function in the static limit.
Returns:¶
- eps0: float
Dielectric constant without local field corrections.
- eps: float
Dielectric constant with local field correction. (RPA, ALDA)
- get_polarizability(q_c=[0, 0, 0], direction='x', filename='polarizability.csv', **xckwargs)[source]¶
Calculate the macroscopic polarizability.
Generate a file ‘polarizability.csv’, unless filename is set to None.
Returns:¶
- alpha0_w: np.ndarray
Polarizability calculated without local-field corrections
- alpha_w: np.ndarray
Polarizability calculated with local-field corrections.