Frequently Asked Questions¶
General¶
Citation: How should I cite GPAW?¶
If you find GPAW useful in your research please cite this GPAW review:
Jens Jørgen Mortensen, Ask Hjorth Larsen, Mikael Kuisma et al.J. Chem. Phys. 160, 092503 (2024)
together with the ASE review (see How should I cite ASE?).
You are welcome to cite also the original GPAW reference and an earlier GPAW review:
J. J. Mortensen, L. B. Hansen, and K. W. JacobsenPhys. Rev. B 71, 035109 (2005)J. Enkovaara, C. Rostgaard, J. J. Mortensen et al.J. Phys.: Condens. Matter 22, 253202 (2010)
Please also cite those of the following that are relevant to you work:
Libxc for XC-functionals other than LDA, PBE, revPBE, RPBE and PW91:
S. Lehtola, C. Steigemann, M. J. T. Oliveira and M. A. L. Marques., Recent developments in LIBXC — a comprehensive library of functionals for density functional theory, SoftwareX 7, 1 (2018)
Time-propagation TDDFT or Linear response TDDFT:
M. Walter, H. Häkkinen, L. Lehtovaara, M. Puska, J. Enkovaara, C. Rostgaard and J. J. Mortensen, Time-dependent density-functional theory in the projector augmented-wave method, J. Chem. Phys. 128, 244101 (2008)
Localized basis set calculations (LCAO):
A. H. Larsen, M. Vanin, J. J. Mortensen, K. S. Thygesen, and K. W. Jacobsen, Localized atomic basis set in the projector augmented wave method, Phys. Rev. B 80, 195112 (2009)
Linear dielectric response of an extended systems:
J. Yan, J. J. Mortensen, K. W. Jacobsen, and K. S. Thygesen, Linear density response function in the projector augmented wave method: Applications to solids, surfaces, and interfaces, Phys. Rev. B 83, 245122 (2011)
Quasi-particle spectrum in the GW approximation:
F. Hüser, T. Olsen, and K. S. Thygesen, Quasiparticle GW calculations for solids, molecules, and two-dimensional materials, Phys. Rev. B 87, 235132 (2013)
Continuum Solvent Model (CSM):
A. Held and M. Walter, Simplified continuum solvent model with a smooth cavity based on volumetric data, J. Chem. Phys. 141, 174108 (2014)
Time-propagation TDDFT with LCAO:
M. Kuisma, A. Sakko, T. P. Rossi, A. H. Larsen, J. Enkovaara, L. Lehtovaara, and T. T. Rantala, Localized surface plasmon resonance in silver nanoparticles: Atomistic first-principles time-dependent density functional theory calculations, Phys. Rev. B 91, 115431 (2015)
Kohn–Sham decomposition of density matrix and Time-propagation TDDFT with LCAO:
T. P. Rossi, M. Kuisma, M. J. Puska, R. M. Nieminen, and P. Erhart, Kohn–Sham Decomposition in Real-Time Time-Dependent Density-Functional Theory: An Efficient Tool for Analyzing Plasmonic Excitations, J. Chem. Theory Comput. 13, 4779 (2017)
Solvated Jellium (constant-potential electrochemistry):
G. Kastlunger, P. Lindgren, A.A. Peterson, Controlled-potential simulation of elementary electrochemical reactions: proton discharge on metal surfaces, J. Phys. Chem. C 122, 12771 (2018)
Citations of the GPAW method papers¶
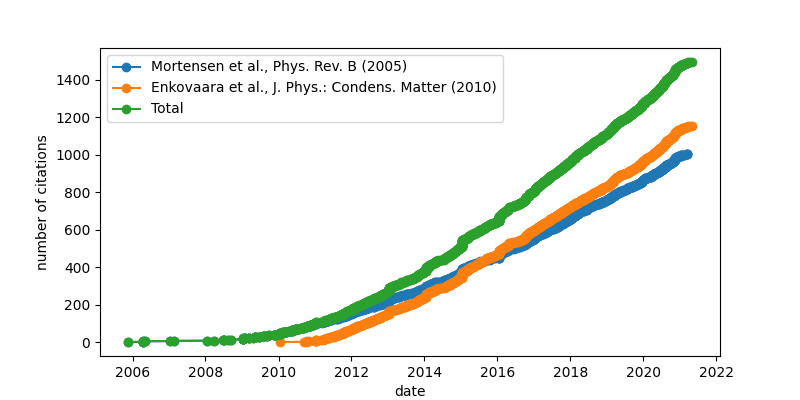
(updated on 18 Mar 2021)
The total number of citations above is the number of publications citing at least one of the other papers, not the sum of all citation counts.
BibTex (doc/GPAW.bib):
@article{Mortensen2005,
author = {Mortensen, J. J. and Hansen, L. B. and Jacobsen, K. W.},
title = {Real-space grid implementation of the projector augmented wave method},
journal = {Phys. Rev. B},
volume = {71},
number = {3},
pages = {035109},
year = {2005},
doi = {10.1103/PhysRevB.71.035109}
}
@article{Enkovaara2010,
author = {Enkovaara, J. and Rostgaard, C. and Mortensen, J. J. and
Chen, J. and Du{\l}ak, M. and Ferrighi, L. and
Gavnholt, J. and Glinsvad, C. and Haikola, V. and
Hansen, H. A. and Kristoffersen, H. H. and Kuisma, M. and
Larsen, A. H. and Lehtovaara, L. and Ljungberg, M. and
Lopez-Acevedo, O. and Moses, P. G. and Ojanen, J. and
Olsen, T. and Petzold, V. and Romero, N. A. and
Stausholm-M{\o}ller, J. and Strange, M. and
Tritsaris, G. A. and Vanin, M. and Walter, M. and
Hammer, B. and H{\"a}kkinen, H. and Madsen, G. K. H. and
Nieminen, R. M. and N{\o}rskov, J. K. and Puska, M. and
Rantala, T. T. and Schi{\o}tz, J. and Thygesen, K. S. and
Jacobsen, K. W.},
title = {Electronic structure calculations with {GPAW}: a real-space implementation of the projector augmented-wave method},
journal = {J. Phys.: Condens. Matter},
volume = {22},
number = {25},
pages = {253202},
year = {2010},
doi = {10.1088/0953-8984/22/25/253202}
}
@article{Lehtola2018,
author = {Susi Lehtola and Conrad Steigemann and Micael
J. T. Oliveira and Miguel A. L. Marques},
title = {Recent developments in libxc -- A comprehensive library of functionals for density functional theory},
journal = {SoftwareX},
volume = {7},
pages = {1-5},
year = {2018},
issn = {2352-7110},
url = {https://www.sciencedirect.com/science/article/pii/S2352711017300602},
keywords = {Density functional theory, Exchange–correlation, Local
density approximations, Generalized gradient
approximations, meta-GGA approximations},
abstract = {libxc is a library of exchange–correlation functionals
for density-functional theory. We are concerned with
semi-local functionals (or the semi-local part of
hybrid functionals), namely local-density
approximations, generalized-gradient approximations,
and meta-generalized-gradient
approximations. Currently we include around 400
functionals for the exchange, correlation, and the
kinetic energy, spanning more than 50 years of
research. Moreover, libxc is by now used by more
than 20 codes, not only from the atomic, molecular,
and solid-state physics, but also from the quantum
chemistry communities.},
doi = {10.1016/j.softx.2017.11.002}
}
@article{Walter2008,
author = {Walter, Michael and H{\"a}kkinen, Hannu and Lehtovaara, Lauri and
Puska, Martti and Enkovaara, Jussi and Rostgaard, Carsten and
Mortensen, Jens J{\o}rgen},
title = {Time-dependent density-functional theory in the projector augmented-wave method},
journal = {J. Chem. Phys.},
volume = {128},
number = {24},
pages = {244101},
year = {2008},
doi = {10.1063/1.2943138}
}
@article{Larsen2009,
author = {Larsen, A. H. and Vanin, M. and Mortensen, J. J. and
Thygesen, K. S. and Jacobsen, K. W.},
title = {Localized atomic basis set in the projector augmented wave method},
journal = {Phys. Rev. B},
volume = {80},
number = {19},
pages = {195112},
year = {2009},
doi = {10.1103/PhysRevB.80.195112}
}
@article{Yan2011,
author = {Yan, Jun and Mortensen, Jens J. and Jacobsen, Karsten W. and
Thygesen, Kristian S.},
title = {Linear density response function in the projector augmented wave method: Applications to solids, surfaces, and interfaces},
journal = {Phys. Rev. B},
volume = {83},
number = {24},
pages = {245122},
year = {2011},
doi = {10.1103/PhysRevB.83.245122}
}
@article{Huser2013,
author = {H\"user, Falco and Olsen, Thomas and Thygesen, Kristian S.},
title = {Quasiparticle GW calculations for solids, molecules, and two-dimensional materials},
journal = {Phys. Rev. B},
volume = {87},
number = {23},
pages = {235132},
year = {2013},
doi = {10.1103/PhysRevB.87.235132}
}
@article{Held2014,
author = {Held, Alexander and Walter, Michael},
title = {Simplified continuum solvent model with a smooth cavity based on volumetric data},
journal = {J. Chem. Phys.},
volume = {141},
number = {17},
pages = {174108},
year = {2014},
doi = {10.1063/1.4900838}
}
@article{Kuisma2015,
author = {Kuisma, M. and Sakko, A. and Rossi, T. P. and Larsen, A. H. and Enkovaara, J. and Lehtovaara, L. and Rantala, T. T.},
title = {Localized surface plasmon resonance in silver nanoparticles: Atomistic first-principles time-dependent density-functional theory calculations},
journal = {Phys. Rev. B},
volume = {91},
number = {11},
pages = {115431},
year = {2015},
doi = {10.1103/PhysRevB.91.115431}
}
@article{Rossi2017,
author = {Rossi, Tuomas P. and Kuisma, Mikael and Puska, Martti J. and
Nieminen, Risto M. and Erhart, Paul},
title = {Kohn--Sham Decomposition in Real-Time Time-Dependent Density-Functional Theory: An Efficient Tool for Analyzing Plasmonic Excitations},
journal = {J. Chem. Theory Comput.},
volume = {13},
number = {10},
pages = {4779-4790},
year = {2017},
doi = {10.1021/acs.jctc.7b00589}
}
@article{Kastlunger2018,
author = {Kastlunger, Georg and Lindgren, Per and Peterson, Andrew A.},
title = {Controlled-Potential Simulation of Elementary Electrochemical Reactions: Proton Discharge on Metal Surfaces},
journal = {The Journal of Physical Chemistry C},
volume = {122},
number = {24},
pages = {12771-12781},
year = 2018,
doi = {10.1021/acs.jpcc.8b02465},
}
How do you pronounce GPAW?¶
In English: “geepaw” with a long “a”.
In Danish: Først bogstavet “g”, derefter “pav”: “g-pav”.
In Finnish: supisuomalaisittain “kee-pav”.
Compiling the C-code¶
For architecture dependent settings see the Platforms and architectures page.
Compilation of the C part failed:
[~]$ python2.4 setup.py build_ext
building '_gpaw' extension
pgcc -fno-strict-aliasing -DNDEBUG -O2 -g -pipe -Wp,-D_FORTIFY_SOURCE=2 -fexceptions -m64 -D_GNU_SOURCE -fPIC -fPIC -I/usr/include/python2.4 -c c/localized_functions.c -o build/temp.linux-x86_64-2.4/c/localized_functions.o -Wall -std=c99
pgcc-Warning-Unknown switch: -fno-strict-aliasing
PGC-S-0040-Illegal use of symbol, _Complex (/usr/include/bits/cmathcalls.h: 54)
You are probably using another compiler, than was used for compiling python. Undefine the environment variables CC, CFLAGS and LDFLAGS with:
# sh/bash users:
unset CC; unset CFLAGS; unset LDFLAGS
# csh/tcsh users:
unsetenv CC; unsetenv CFLAGS; unsetenv LDFLAGS
and try again.
Calculation does not converge¶
Consult the Convergence Issues page.
Poisson solver did not converge!¶
If you are doing a spin-polarized calculation for an isolated molecule, then you should set the Fermi temperature to a low value.
You can also try to set the number of grid points to be divisible by 8. Consult the Notes on performance page.