Exact exchange¶
Currently we have two implementations of exact exchange:
Plane-wave mode implementation: . Handles k-points, exploits symmetries and calculates forces.
Finite-difference mode implementation: hybrid.py. Can handle Gamma-point only calculations self-consistently (for molecules and large cells). No forces.
Self-consistent plane-wave implementation¶
The following values for the xc names can be used: EXX, PBE0,
HSE03, HSE06 and B3LYP. Here is an example for a PW-mode
HSE06 calculation:
atoms.calc = GPAW(
mode={'name': 'pw', 'ecut': ...},
xc='HSE06',
...)
or equivalently for full controll:
atoms.calc = GPAW(
mode={'name': 'pw', 'ecut': ...},
xc={'name': 'HYB_GGA_XC_HSE06',
'fraction': 0.25,
'omega': 0.11,
'backend': 'pw'},
...)
Note
At the moment one must use xc={'name': ..., 'backend': 'pw'} for
EXX, PBE0 and B3LYP!
Non self-consistent plane-wave implementation¶
- gpaw.hybrids.energy.non_self_consistent_energy(calc, xcname, ftol=1e-09)[source]¶
Calculate non self-consistent energy for Hybrid functional.
Based on a self-consistent DFT calculation (calc). EXX integrals involving states with occupation numbers less than ftol are skipped.
>>> energies = non_self_consistent_energy('<gpw-file>', ... xcname='HSE06') >>> e_hyb = energies.sum()
The correction to the self-consistent energy will be
energies[1:].sum().The returned energy contributions are (in eV):
DFT total free energy (not extrapolated to zero smearing)
minus DFT XC energy
Hybrid semi-local XC energy
EXX core-core energy
EXX core-valence energy
EXX valence-valence energy
- gpaw.hybrids.eigenvalues.non_self_consistent_eigenvalues(calc, xcname, n1=0, n2=0, kpt_indices=None, snapshot=None, ftol=1e-09)[source]¶
Calculate non self-consistent eigenvalues for a hybrid functional.
Based on a self-consistent DFT calculation (calc). Only eigenvalues n1 to n2 - 1 for the IBZ indices in kpt_indices are calculated (default is all bands and all k-points). EXX integrals involving states with occupation numbers less than ftol are skipped. Use snapshot=’name.json’ to get snapshots for each k-point finished.
Returns three (nspins, nkpts, n2 - n1)-shaped ndarrays with contributions to the eigenvalues in eV:
>>> nsceigs = non_self_consistent_eigenvalues >>> eig_dft, vxc_dft, vxc_hyb = nsceigs('<gpw-file>', xcname='PBE0') >>> eig_hyb = eig_dft - vxc_dft + vxc_hyb
See this tutorial: PBE0 calculations for bulk silicon.
Here is an example:
"""EXX hydrogen atom.
Compare self-consistent EXX calculation with non self-consistent
EXX calculation on top of LDA.
"""
from ase import Atoms
from ase.units import Ry
from gpaw import GPAW, PW
from gpaw.hybrids.eigenvalues import non_self_consistent_eigenvalues
from gpaw.hybrids.energy import non_self_consistent_energy
atoms = Atoms('H', magmoms=[1.0])
atoms.center(vacuum=5.0)
# Self-consistent calculation:
atoms.calc = GPAW(mode=PW(600),
xc='EXX:backend=pw')
eexx = atoms.get_potential_energy() + atoms.calc.get_reference_energy()
# Check energy
eexxref = -1.0 * Ry
assert abs(eexx - eexxref) < 0.001
# ... and eigenvalues
eig1, eig2 = (atoms.calc.get_eigenvalues(spin=spin)[0] for spin in [0, 1])
eigref1 = -1.0 * Ry
eigref2 = ... # ?
assert abs(eig1 - eigref1) < 0.03
# assert abs(eig2 - eigref2) < 0.03
# LDA:
atoms.calc = GPAW(mode=PW(600),
xc='LDA')
atoms.get_potential_energy()
# Check non self-consistent eigenvalues
result = non_self_consistent_eigenvalues(atoms.calc,
'EXX',
snapshot='h-hse-snapshot.json')
eiglda, vlda, vexx = result
eig1b, eig2b = (eiglda - vlda + vexx)[:, 0, 0]
assert abs(eig1b - eig1) < 0.04
assert abs(eig2b - eig2) < 1.1
# ... and energy
energies = non_self_consistent_energy(atoms.calc, 'EXX')
eexxb = energies.sum() + atoms.calc.get_reference_energy()
assert abs(eexxb - eexx) < 0.03
Self-consistent finite-difference implementation¶
The current implementation lacks the following features:
Support for periodic systems. Actually, the code won’t complain, but the results have not been tested.
Support for k-point sampling. No consideration has been made as to multiple k-points, or even comlplex wave functions, so this definitely won’t work.
Forces. Force evaluations when including (a fraction of -) the fock operator in the xc-functional has been implemented, but has not been tested.
Speed. Inclusion of Fock exchange is exceedingly slow. The bottleneck is solving the poisson integrals of the Fock operator, which is currently done using an iterative real-space solver with a zero initial guess for the potential at each SCF cycle. This should be optimized.
One way to speed up an exact-exchange (or hybrid) calculation is to use the
coarse grid (used for wave functions) instead of the finegrid (used for for
densities) for the Fock potentials. This should give a speed-up factor of 8.
This can be specified in the xc keyword like in this example
test_coarse.py
Parallelization using domain decomposition is fully supported.
The Fock operator can be used to do the hybrid functional PBE0, and of course,
Hartree-Fock type EXX. These are accessed by setting the xc keyword to
PBE0 or EXX respectively.
The following functionals are suppported:
Functional |
Type |
Reference |
|---|---|---|
EXX |
Global |
|
PBE0 |
Global |
|
B3LYP |
Global |
|
CAMY-B3LYP |
RSF-LR |
|
CAMY-BLYP |
RSF-LR |
|
CAMY-B3LYP |
RSF-LR |
|
LCY-BLYP |
RSF-LR |
|
LCY-PBE |
RSF-LR |
Here “Global” denotes global hybrids, which use the same percentage of Hartree-Fock exchange for every point in space, while “RSF-SR” and “RSF-LR” denotes range-separated functionals which mix the fraction of Hartree-Fock and DFT exchange based on the spatial distance between two points, where for a “RSF-SR” the amount of Hartree-Fock exchange decrease with the distance and increase for a “RSF-LR”. See Range separated functionals (RSF) for more detailed information on RSF(-LR).
A thesis on the implementation of EXX in the PAW framework, and the specifics of the GPAW project can be seen on the reports presentations and theses page.
A comparison of the atomization energies of the g2-1 test-set calculated in VASP, Gaussian03, and GPAW is shown in the below two figures for the PBE and the PBE0 functional respectively.
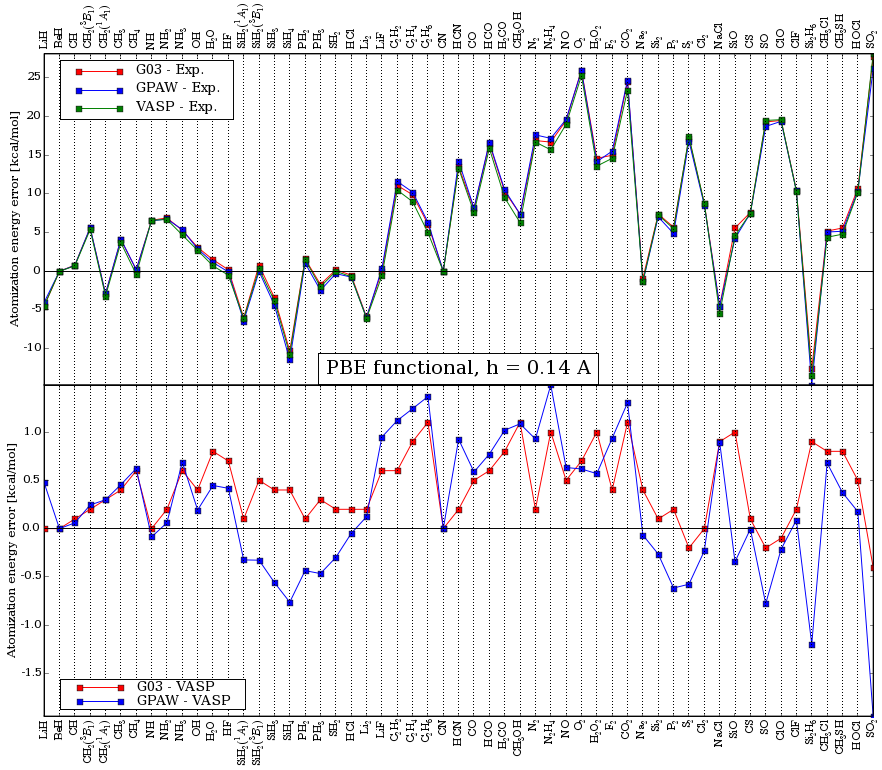
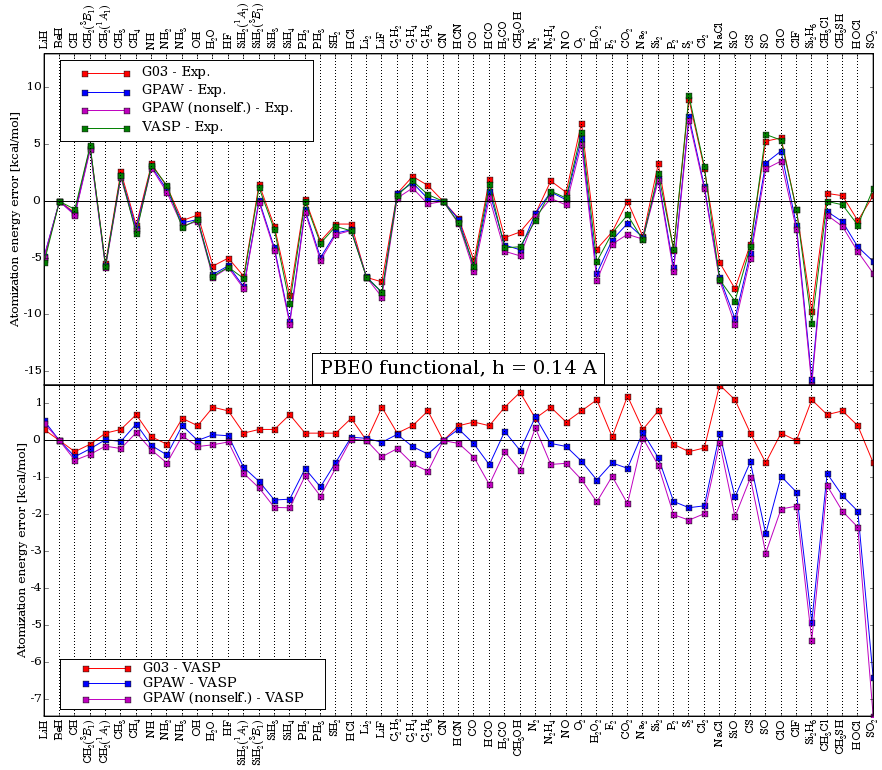
In the last figure, the curve marked GPAW (nonself.) is a non
self-consistent PBE0 calculation using self-consistent PBE orbitals.
It should be noted, that the implementation lacks an optimized effective
potential. Therefore the unoccupied states utilizing EXX as implemented in
GPAW usually approximate (excited) electron affinities. Therefore calculations
utilizing Hartree-Fock exchange are usually a bad basis for the calculation of
optical excitations by lrTDDFT. As a remedy, the improved virtual orbitals
(IVOs, [HA71]) were implemented. The requested excitation basis can be chosen
by the keyword excitation and the state by excited where the state is
counted from the HOMO downwards:
"""Calculate the excitation energy of NaCl by an RSF using IVOs."""
from ase.build import molecule
from ase.units import Hartree
from gpaw import GPAW, setup_paths
from gpaw.mpi import world
from gpaw.occupations import FermiDirac
from gpaw.test import gen
from gpaw.eigensolvers import RMMDIIS
from gpaw.utilities.adjust_cell import adjust_cell
from gpaw.lrtddft import LrTDDFT
h = 0.3 # Gridspacing
e_singlet = 4.3
e_singlet_lr = 4.3
if setup_paths[0] != '.':
setup_paths.insert(0, '.')
gen('Na', xcname='PBE', scalarrel=True, exx=True, yukawa_gamma=0.40)
gen('Cl', xcname='PBE', scalarrel=True, exx=True, yukawa_gamma=0.40)
c = {'energy': 0.005, 'eigenstates': 1e-2, 'density': 1e-2}
mol = molecule('NaCl')
adjust_cell(mol, 5.0, h=h)
calc = GPAW(mode='fd', txt='NaCl.txt',
xc='LCY-PBE:omega=0.40:excitation=singlet',
eigensolver=RMMDIIS(), h=h, occupations=FermiDirac(width=0.0),
spinpol=False, convergence=c)
mol.calc = calc
mol.get_potential_energy()
(eps_homo, eps_lumo) = calc.get_homo_lumo()
e_ex = eps_lumo - eps_homo
assert abs(e_singlet - e_ex) < 0.15
calc.write('NaCl.gpw')
lr = LrTDDFT(calc, txt='LCY_TDDFT_NaCl.log',
restrict={'istart': 6, 'jend': 7}, nspins=2)
lr.write('LCY_TDDFT_NaCl.ex.gz')
if world.rank == 0:
lr2 = LrTDDFT.read('LCY_TDDFT_NaCl.ex.gz')
lr2.diagonalize()
ex_lr = lr2[1].get_energy() * Hartree
assert abs(e_singlet_lr - e_singlet) < 0.05
Support for IVOs in lrTDDFT is done along the work of Berman and Kaldor [BK79].
If the number of bands in the calculation exceeds the number of bands delivered by the datasets, GPAW initializes the missing bands randomly. Calculations utilizing Hartree-Fock exchange can only use the RMM-DIIS eigensolver. Therefore the states might not converge to the energetically lowest states. To circumvent this problem on can made a calculation using a semi-local functional like PBE and uses this wave-functions as a basis for the following calculation utilizing Hartree-Fock exchange as shown in the following code snippet which uses PBE0 in conjuncture with the IVOs:
"""Test calculation for unoccupied states using IVOs."""
from ase.build import molecule
from gpaw.utilities.adjust_cell import adjust_cell
from gpaw import GPAW, KohnShamConvergenceError, FermiDirac
from gpaw.eigensolvers import CG, RMMDIIS
calc_parms = [
{'xc': 'PBE0:unocc=True',
'eigensolver': RMMDIIS(niter=5),
'convergence': {
'energy': 0.005,
'bands': -2,
'eigenstates': 1e-4,
'density': 1e-3}},
{'xc': 'PBE0:excitation=singlet',
'convergence': {
'energy': 0.005,
'bands': 'occupied',
'eigenstates': 1e-4,
'density': 1e-3}}]
def calc_me(atoms, nbands):
"""Do the calculation."""
molecule_name = atoms.get_chemical_formula()
atoms.set_initial_magnetic_moments([-1.0, 1.0])
fname = '.'.join([molecule_name, 'PBE-SIN'])
calc = GPAW(mode='fd',
h=0.25,
xc='PBE',
eigensolver=CG(niter=5),
nbands=nbands,
txt=fname + '.log',
occupations=FermiDirac(0.0, fixmagmom=True),
convergence={
'energy': 0.005,
'bands': nbands,
'eigenstates': 1e-4,
'density': 1e-3})
atoms.calc = calc
try:
atoms.get_potential_energy()
except KohnShamConvergenceError:
pass
if calc.scf.converged:
for calcp in calc_parms:
calc.set(**calcp)
try:
calc.calculate(system_changes=[])
except KohnShamConvergenceError:
break
if calc.scf.converged:
calc.write(fname + '.gpw', mode='all')
loa = molecule('NaCl')
adjust_cell(loa, border=6.0, h=0.25, multiple=16)
loa.center()
loa.translate([0.001, 0.002, 0.003])
nbands = 25
calc_me(loa, nbands)
C. Adamo and V. Barone. Toward Chemical Accuracy in the Computation of NMR Shieldings: The PBE0 Model.. Chem. Phys. Lett. 298.1 (11. Dec. 1998), S. 113–119.
V. Barone. Inclusion of Hartree–Fock exchange in density functional methods. Hyperfine structure of second row atoms and hydrides. Jour. Chem. Phys. 101.8 (1994), S. 6834–6838.
M. Berman and U. Kaldor. Fast calculation of excited-state potentials for rare-gas diatomic molecules: Ne2 and Ar2. Chem. Phys. 43.3 (1979), S. 375–383.
S. Huzinaga and C. Arnau. Virtual Orbitals in Hartree–Fock Theory. II. Jour. Chem. Phys. 54.5 (1. Ma. 1971), S. 1948–1951.