PBE0 calculations for bulk silicon¶
This tutorial will do non self-consistent PBE0 based on self-consistent PBE.
See also
Band structure exercice.
PBE and PBE0 band gaps¶
The band structure can be calculated like this:
# web-page: si-gaps.csv
from ase.build import bulk
from ase.parallel import paropen
from gpaw.hybrids.eigenvalues import non_self_consistent_eigenvalues
from gpaw import GPAW, PW
a = 5.43
si = bulk('Si', 'diamond', a)
fd = paropen('si-gaps.csv', 'w')
for k in range(2, 9, 2):
name = f'Si-{k}'
si.calc = GPAW(kpts={'size': (k, k, k), 'gamma': True},
mode=PW(200),
xc='PBE',
convergence={'bands': 5},
txt=name + '.txt')
si.get_potential_energy()
si.calc.write(name + '.gpw', mode='all')
# Range of eigenvalues:
n1 = 3
n2 = 5
ibzkpts = si.calc.get_ibz_k_points()
kpt_indices = []
for kpt in [(0, 0, 0), (0.5, 0.5, 0)]: # Gamma and X
# Find k-point index:
i = abs(ibzkpts - kpt).sum(1).argmin()
kpt_indices.append(i)
# Do PBE0 calculation:
epbe, vpbe, vpbe0 = non_self_consistent_eigenvalues(
name + '.gpw',
'PBE0',
n1, n2,
kpt_indices,
snapshot=name + '.json')
epbe0 = epbe - vpbe + vpbe0
gg = epbe[0, 0, 1] - epbe[0, 0, 0]
gx = epbe[0, 1, 1] - epbe[0, 0, 0]
gg0 = epbe0[0, 0, 1] - epbe0[0, 0, 0]
gx0 = epbe0[0, 1, 1] - epbe0[0, 0, 0]
print(f'{k}, {gg:.3f}, {gx:.3f}, {gg0:.3f}, {gx0:.3f}', file=fd)
fd.flush()
assert abs(gg - 2.559) < 0.01
assert abs(gx - 0.707) < 0.01
assert abs(gg0 - 3.873) < 0.01
assert abs(gx0 - 1.828) < 0.01
These are the resulting \(\Gamma\)-\(\Gamma\) and \(\Gamma\)-\(X\) gaps for PBE and PBE0 in eV:
k-points |
PBE(G-G) |
PBE(G-X) |
PBE0(G-G) |
PBE0(G-X) |
|---|---|---|---|---|
2 |
2.445 |
0.598 |
4.187 |
2.153 |
4 |
2.538 |
0.688 |
3.954 |
1.913 |
6 |
2.553 |
0.701 |
3.892 |
1.849 |
8 |
2.555 |
0.704 |
3.874 |
1.831 |
Lattice constant and bulk modulus¶
Here is how to calculate the lattice constant:
import ase.db
from ase.build import bulk
import numpy as np
from gpaw.hybrids.energy import non_self_consistent_energy as nsc_energy
from gpaw import GPAW, PW
a0 = 5.43
con = ase.db.connect('si.db')
for k in range(2, 9):
for a in np.linspace(a0 - 0.04, a0 + 0.04, 5):
id = con.reserve(a=a, k=k)
if id is None:
continue
si = bulk('Si', 'diamond', a)
si.calc = GPAW(kpts=(k, k, k),
mode=PW(400),
xc='PBE',
eigensolver='rmm-diis',
txt=None)
si.get_potential_energy()
name = f'si-{a:.2f}-{k}'
si.calc.write(name + '.gpw', mode='all')
epbe0 = nsc_energy(name + '.gpw', 'PBE0').sum()
con.write(si, a=a, k=k, epbe0=epbe0)
del con[id]
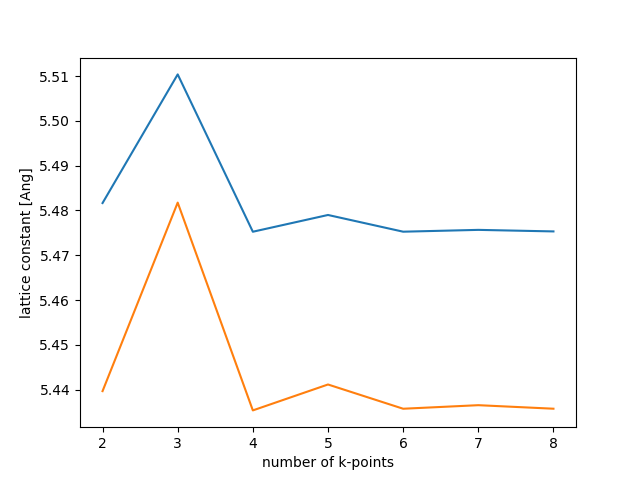
Plot the results like this:
# web-page: si-a.png
import matplotlib.pyplot as plt
import ase.db
from ase.eos import EquationOfState
def lattice_constant(volumes, energies):
eos = EquationOfState(volumes, energies)
v, e, B = eos.fit()
a = (v * 4)**(1 / 3)
return a
con = ase.db.connect('si.db')
results = []
K = list(range(2, 9))
A = []
A0 = []
for k in K:
rows = list(con.select(k=k))
V = [row.volume for row in rows]
E = [row.energy for row in rows]
E0 = [row.epbe0 for row in rows]
A.append(lattice_constant(V, E))
A0.append(lattice_constant(V, E0))
print(K, A, A0)
plt.plot(K, A, label='PBE')
plt.plot(K, A0, label='PBE0')
plt.xlabel('number of k-points')
plt.ylabel('lattice constant [Ang]')
plt.savefig('si-a.png')