Occupation number smearing¶
See also
Convergence with respect to number of k-point for bulk Cu energy with different smearing methods:
from ase.build import bulk
from gpaw import GPAW, PW
cu = bulk('Cu', 'fcc', a=3.6)
for smearing in [{'name': 'improved-tetrahedron-method'},
{'name': 'tetrahedron-method'},
{'name': 'fermi-dirac', 'width': 0.05},
{'name': 'marzari-vanderbilt', 'width': 0.2}]:
name = ''.join(word[0].upper() for word in smearing['name'].split('-'))
width = smearing.get('width')
if width:
name += f'-{width}'
for k in range(8, 21):
cu.calc = GPAW(
mode=PW(400),
kpts=(k, k, k),
occupations=smearing,
txt=f'Cu-{name}-{k}.txt')
e = cu.get_potential_energy()
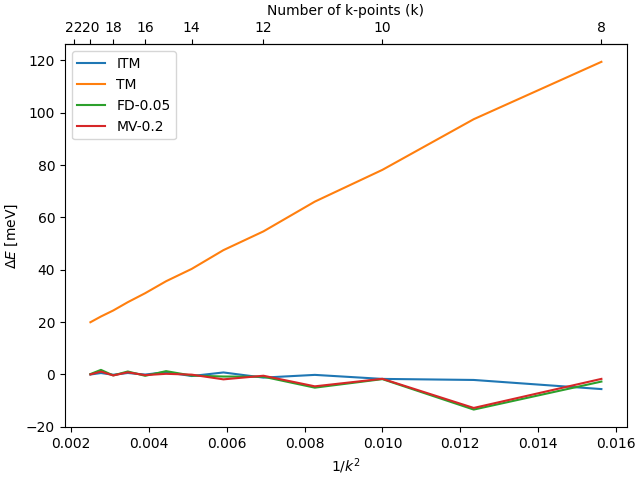
(made with cu_plot.py). See also figure 3 in
Blöchl et. al.
- gpaw.occupations.create_occ_calc(dct, *, parallel_layout=None, fixed_magmom_value=None, rcell=None, monkhorst_pack_size=None, bz2ibzmap=None, nspins=None, nelectrons=None, nkpts=None, nbands=None)[source]¶
Surprise: Create occupation-number object.
The unit of width is eV and name must be one of:
‘fermi-dirac’
‘marzari-vanderbilt’
‘methfessel-paxton’
‘fixed’
‘tetrahedron-method’
‘improved-tetrahedron-method’
‘orbital-free’
>>> occ = create_occ_calc({'width': 0.0}) >>> occ.calculate(nelectrons=3, ... eigenvalues=[[0, 1, 2], [0, 2, 3]], ... weights=[1, 1]) (array([[1., 1., 0.], [1., 0., 0.]]), [1.5], 0.0)
- gpaw.occupations.fermi_dirac(eig, fermi_level, width)[source]¶
Fermi-Dirac distribution function.
>>> f, _, _ = fermi_dirac(np.array([-1.0, 0.0, 1.0]), 0.0, 0.01) >>> f.round(5) array([1. , 0.5, 0. ])
- gpaw.occupations.marzari_vanderbilt(eig, fermi_level, width)[source]¶
Marzari-Vanderbilt distribution (cold smearing).
See: doi: 10.1103/PhysRevLett.82.3296
- gpaw.occupations.methfessel_paxton(eig, fermi_level, width, order=0)[source]¶
Methfessel-Paxton distribution.
- class gpaw.occupations.OccupationNumberCalculator(parallel_layout=None)[source]¶
Base class for all occupation number calculators.
Object for calculating fermi level(s) and occupation numbers.
If fixmagmom=True then the fixed_magmom_value attribute must be set and two fermi levels will be calculated.
- calculate(nelectrons, eigenvalues, weights, fermi_levels_guess=None)[source]¶
Calculate occupation numbers and fermi level(s) from eigenvalues.
- nelectrons:
Number of electrons.
- eigenvalues: ndarray, shape=(nibzkpts, nbands)
Eigenvalues in Hartree.
- weights: ndarray, shape=(nibzkpts,)
Weights of k-points in IBZ (must sum to 1).
- parallel:
Parallel distribution of eigenvalues.
- fermi_level_guesses:
Optional guess(es) at fermi level(s).
Returns a tuple containing:
occupation numbers (in the range 0 to 1)
fermi-level in Hartree
entropy as -S*T in Hartree
>>> occ = ZeroWidth() >>> occ.calculate(1, [[0, 1]], [1]) (array([[1., 0.]]), [0.5], 0.0)
- class gpaw.occupations.FixedOccupationNumbers(numbers, parallel_layout=None)[source]¶
Fixed occupation numbers.
- f_sn: ndarray, shape=(nspins, nbands)
Occupation numbers (in the range from 0 to 1)
Example (excited state with 4 electrons):
occ = FixedOccupationNumbers([[1, 0, 1, 0], [1, 1, 0, 0]])
- class gpaw.occupations.ParallelLayout(bd, kpt_comm, domain_comm)[source]¶
Collection of parallel stuff.
Create new instance of ParallelLayout(bd, kpt_comm, domain_comm)
- gpaw.occupations.occupation_numbers(occ, eig_skn, weight_k, nelectrons)[source]¶
Calculate occupation numbers from eigenvalues in eV (deprecated).
- occ: dict
Example: {‘name’: ‘fermi-dirac’, ‘width’: 0.05} (width in eV).
- eps_skn: ndarray, shape=(nspins, nibzkpts, nbands)
Eigenvalues.
- weight_k: ndarray, shape=(nibzkpts,)
Weights of k-points in IBZ (must sum to 1).
- nelectrons: int or float
Number of electrons.
Returns a tuple containing:
f_skn (sums to nelectrons)
fermi-level [Hartree]
magnetic moment
entropy as -S*T [Hartree]
Tetrahedron method¶
- class gpaw.tetrahedron.TetrahedronMethod(rcell, size, improved=False, bz2ibzmap=None, parallel_layout=None)[source]¶
Tetrahedron method for calculating occupation numbers.
The reciprocal cell, rcell, can be given in arbitrary units (only the shape matters) and size is the size of the Monkhorst-Pack grid. If k-points have been symmetry-reduced the bz2ibzmap parameter mapping BZ k-point indizes to IBZ k-point indices must be given.