Setup generation¶
The generation of setups, starts from a spin-paired atomic all-electron calculation with spherical symmetry.
Select the states to include as valence.
Add extra unbound states for extra flexibility.
Select cutoff radius (may be \(\ell\)-dependent).
From the set of bound and unbound all-electron partial waves (\(\phi_{n\ell}(r)\)) construct pseudo partial waves (\(\tilde\phi_{n\ell}(r)\)).
This is done by the Command line interface tool gpaw dataset:
usage: gpaw dataset [-h] [-f <XC>] [-C CONFIGURATION]
[-P PROJECTORS] [-r RADIUS]
[-0 nderivs,radius] [-c radius]
[-z type,nderivs] [-p]
[-l spdfg,e1:e2:de,radius] [-w] [-s] [-n]
[-t TAG] [-a ALPHA] [-g GAMMA] [-b]
[--nlcc] [--core-hole CORE_HOLE]
[-e ELECTRONS]
symbol
Type gpaw dataset --help to get started.
Example¶
Generate dataset for nitrogen and check logarithmic derivatives:
$ gpaw dataset N -fPBE -s -P 2s,s,2p,p,d,F -r 1.3 -l spdfg -p
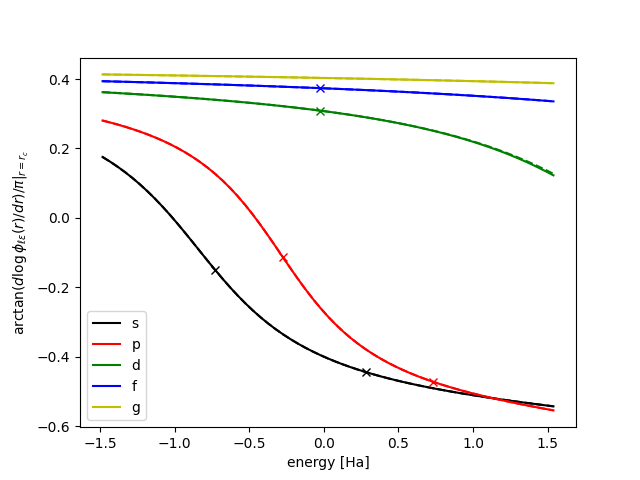
When things look OK, you can write it to a PAW-XML file:
$ gpaw dataset N -fPBE -s -P 2s,s,2p,p,d,F -r 1.3 -t new -w
and use it in you calculations with setups={'N': 'new'}.
See also
The
gpaw atomcommand-line tool for doing an all-electron calculation for an atom
What an OK dataset looks like will depend on what you want:
If you want a very accurate dataset then you will want to include semi-core states and reduce the cutoff radii. Also, you should try to get get correct scattering properties at high energies (check this by looking at the logarithmic derivatives).
If you want something that is computationally efficient then you will want as few valence electrons as possible and as large cutoff radii as possible.
Always do test calculations to compare the performance of your dataset against accurate all-electron reference calculations.
Warning
The gpaw dataset generator (generator2.py)
is work in progress and does not have a stable API. If you manage to
generate a dataset with is that you want to use then hold on to that
file because we can not guarantee that future versions of GPAW will be
able to generate it again.
The Setup releases that we distribute are all generated with the
old gpaw-setup generator (see more below).
Using your own setups¶
The setups you generate must be placed in a directory which is included in
the environment variable GPAW_SETUP_PATH in order for GPAW to
find them. If you want to use the setups in your local directory, add the
following lines to the beginning of your Python script:
from gpaw import setup_paths
setup_paths.insert(0, '.')
You can also override the environment variable GPAW_SETUP_PATH so
that it lists the local directory first and the regular entries afterwards.
If you use bash, GPAW_SETUP_PATH can be temporarily modified
while you run GPAW with the single command:
GPAW_SETUP_PATH=.:$GPAW_SETUP_PATH python3 script.py
or if you are using csh or tcsh, you have to first run setenv and then
GPAW:
setenv GPAW_SETUP_PATH .:$GPAW_SETUP_PATH&& python3 script.py
Old generator¶
The following parameters define a setup:
name |
description |
example |
|---|---|---|
|
Froze core |
|
|
Cutoff radius/radii for projector functions |
|
|
Extra non-bound projectors |
|
|
Zero-potential |
|
|
Fourier-filtering parameters |
|
|
Cutoff radius for compensation charges |
|
The default (LDA) sodium setup can be generated with the command gpaw-setup
Na, which will use default parameters from the file
gpaw/atom/generator.py. See Exchange-Correlation functional for other functionals.